
Mitogenome of the Doleschallia bisaltide and Phylogenetic Analysis of Nymphalinae (Lepidoptera, Nymphalidae)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling, Identification and DNA Extraction
2.2. Nuclear DNA Amplification and Sequencing
2.3. Mitogenome Sequencing, Annotation and Analyses
2.4. Dataset Partitioning and Model Selection
2.5. Phylogenetic Inference
3. Results and Discussion
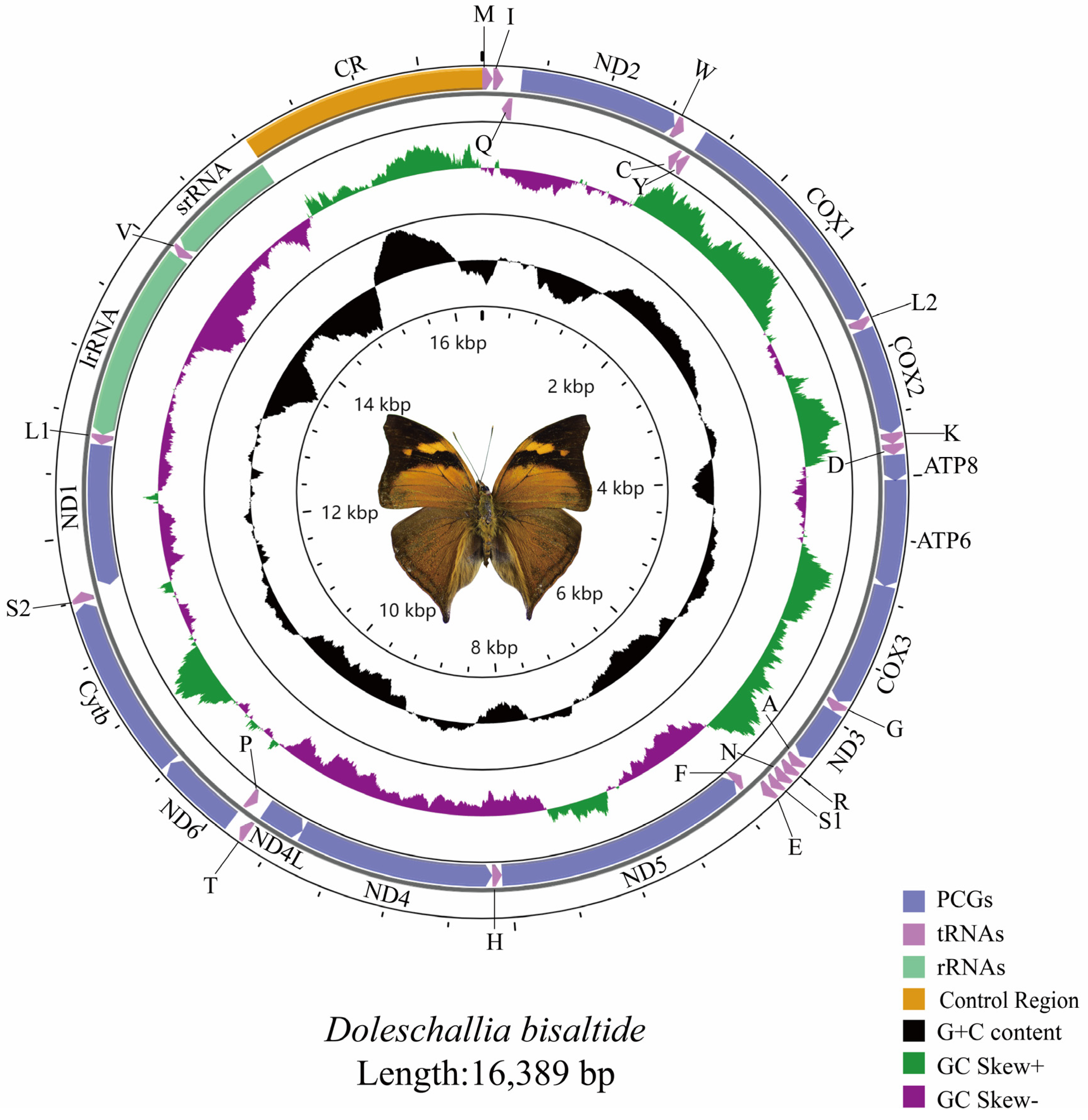
3.1. Basic Characteristics of the Mitochondrial Genome
3.2. Protein-coding Genes, tRNAs, rRNAs and Control Region
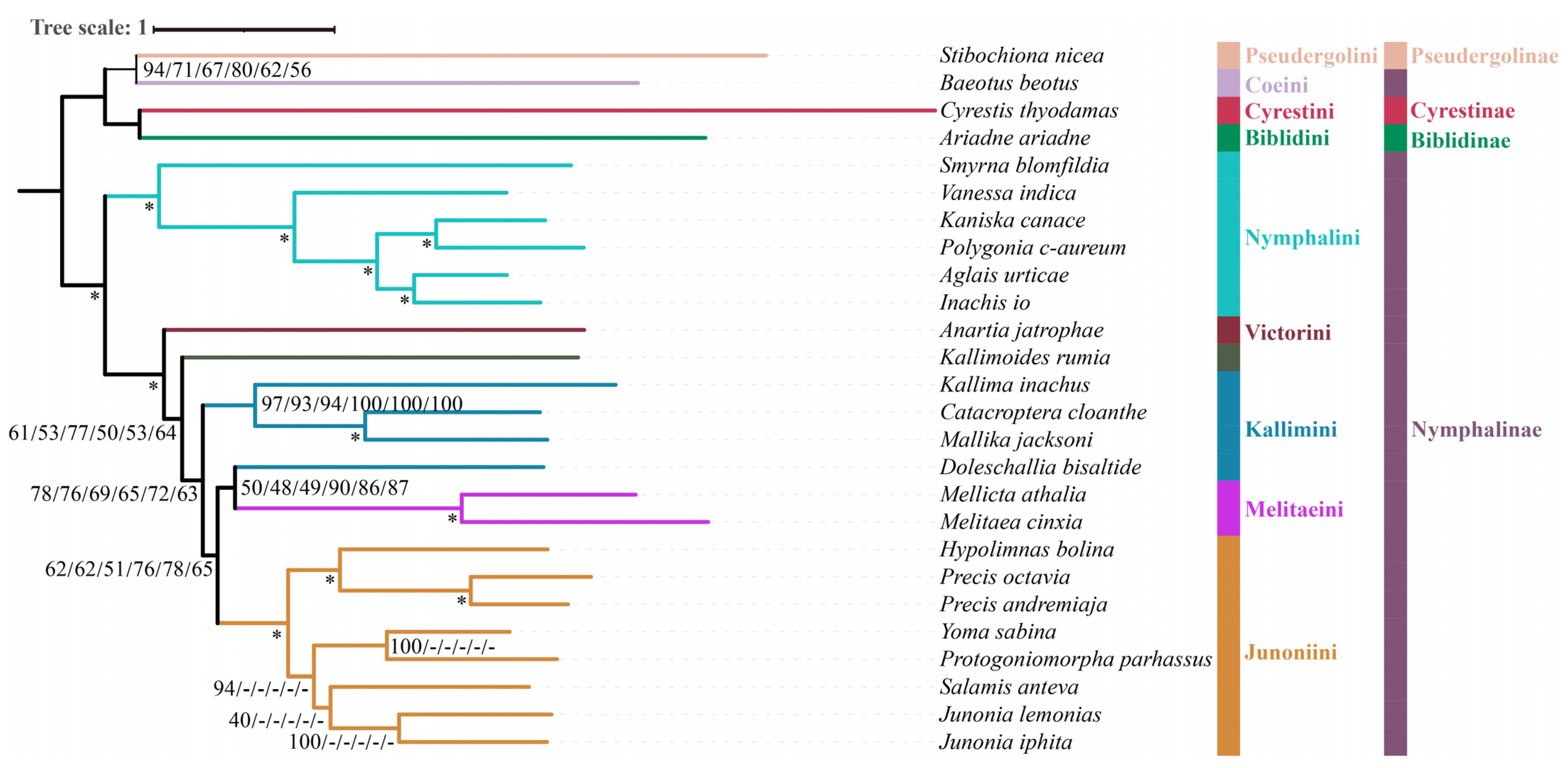
3.3. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wahlberg, N.; Brower, A.V.Z.; Nylin, S. Phylogenetic relationships and historical biogeography of tribes and genera in the subfamily Nymphalinae (Lepidoptera: Nymphalidae). Biol. J. Linn. Soc. 2005, 86, 227–251. [Google Scholar] [CrossRef]
- Boggs, C.L.; Watt, W.B.; Ehrlich, P.R. Butterflies—Ecology and evolution—Taking flight. Science 2004, 303, 174. [Google Scholar]
- Roe, A.D.; Weller, S.J.; Baixeras, J.; Brown, J.; Cummings, M.P.; Davis, D.R.; Kawahara, A.Y.; Parr, C.S.; Regier, J.C.; Rubinoff, D.; et al. Evolutionary framework for Lepidoptera model systems. In Molecular Biology and Genetics of the Lepidoptera, 1st ed.; Goldsmith, M.R., Marec, F., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 1–24. [Google Scholar]
- Espeland, M.; Breinholt, J.; Willmott, K.R.; Warren, A.D.; Vila, R.; Toussaint, E.F.A.; Maunsell, S.C.; Aduse-Poku, K.; Talavera, G.; Eastwood, R.; et al. A comprehensive and dated phylogenomic analysis of butterflies. Curr. Biol. 2018, 28, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.T.; Teng, D.Q.; Li, X.Y.; Yang, P.W.; Da, W.; Zhang, Y.M.; Zhang, Y.B.; Liu, G.C.; Zhang, X.S.; Wan, W.T.; et al. The evolution and diversification of oakleaf butterflies. Cell 2022, 185, 3138–3152. [Google Scholar] [CrossRef] [PubMed]
- Ackery, P.R. Hostplants and classification: A review of nymphalid butterflies. Biol. J. Linn. Soc. 1988, 33, 95–203. [Google Scholar] [CrossRef]
- Chou, I. Classification and Identification of Chinese Butterflies; Henan Scientific and Technological Publishing House: Zhengzhou, China, 1998. [Google Scholar]
- Chou, I. Monograph of Chinese Butterflies, Second Volume; Henan Scientific and Technological Publishing House: Zhengzhou, China, 1994. [Google Scholar]
- Harvey, D.J. Higher classification of the Nymphalidae, Appendix B. In The Development and Evolution of Butterfly Wing Patterns; Nijhout, H.F., Ed.; Smithsonian Institution Press: Washington, DC, USA, 1991; pp. 255–273. [Google Scholar]
- Ackery, P.R.; de Jong, R.; Vane-Wright, R.I. The Butterflies: Hedyloidea, Hesperioidea and Papilionoidea. In Lepidoptera, Moths and Butterflies; Volume Evolution, Systematics and Biogeography; Kristensen, N.P., Ed.; De Gruyter: Berlin, Germany, 1999; pp. 263–300. [Google Scholar]
- Brower, A.V.Z. Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferred from partial sequences of the wingless gene. Proc. Roy. Soc. B Biol. Sci. 2000, 267, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Weingartner, E.; Nylin, S. Towards a better understanding of the higher systematics of Nymphalidae (Lepidoptera: Papilionoidea). Mol. Phylogenet. Evol. 2003, 28, 473–484. [Google Scholar] [CrossRef]
- Lang, S.Y. The Nymphalidae of China (Lepidoptera, Rhopalocera) Part I; Tshikolovets Publications: Pardubice, Czech Republic, 2012. [Google Scholar]
- Freitas, A.V.L.; Brown, K.S.J. Phylogeny of the Nymphalidae (Lepidoptera). Syst. Biol. 2004, 53, 363–383. [Google Scholar] [CrossRef]
- Zhang, J.; Cong, Q.; Shen, J.; Opler, P.A.; Grishin, N.V. Genomics-guided refinement of butterfly taxonomy. Taxon. Rep. Int. Lepid. Surv. 2021, 9, 1–112. [Google Scholar]
- Wahlberg, N.; Wheat, C.W. Genomic outposts serve the phylogenomic pioneers: Designing novel nuclear markers for genomic DNA extractions of Lepidoptera. Syst. Biol. 2008, 57, 231–242. [Google Scholar] [CrossRef]
- Wahlberg, N.; Leneveu, J.; Kodandaramaiah, U.; Pena, C.; Nylin, S.; Freitas, A.V.L.; Brower, A.V.Z. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. P. Roy. Soc. B Biol. Sci. 2009, 276, 4295–4302. [Google Scholar] [CrossRef]
- Wu, L.W.; Lin, L.H.; Lees, D.C.; Hsu, Y.F. Mitogenomic sequences effectively recover relationships within brush-footed butterflies (Lepidoptera: Nymphalidae). BMC Genomics 2014, 15, 468. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Shi, Q.; Sun, X.; Ma, J.; Li, C.; Hao, J.; Yang, Q. Dated phylogeny and dispersal history of the butterfly subfamily Nymphalinae (Lepidoptera: Nymphalidae). Sci. Rep. 2017, 7, 8799. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, I.; Tanikawa-Dodo, Y.; Saigusa, T.; Nishiyama, T.; Kitani, M.; Hasebe, M.; Mohri, H. Phylogeny, biogeography, and host-plant association in the subfamily Apaturinae (Insecta: Lepidoptera: Nymphalidae) inferred from eight nuclear and seven mitochondrial genes. Mol. Phylogenet. Evol. 2010, 57, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Wahlberg, N.; Weingartner, E.; Kodandaramaiah, U.; Nylin, S.; Freitas, A.V.; Brower, A.V. Higher level phylogeny of Satyrinae butterflies (Lepidoptera: Nymphalidae) based on DNA sequence data. Mol. Phylogenet. Evol. 2006, 40, 29–49. [Google Scholar] [CrossRef]
- Wu, L.-W.; Chiba, H.; Lees, D.C.; Ohshima, Y.; Jeng, M.-L. Unravelling relationships among the shared stripes of sailors: Mitogenomic phylogeny of Limenitidini butterflies (Lepidoptera, Nymphalidae, Limenitidinae), focusing on the genera Athyma and Limenitis. Mol. Phylogenet. Evol. 2019, 130, 60–66. [Google Scholar] [CrossRef]
- Wu, C.S.; Hsu, Y.F. Butterflies of China; The Straits Publishing House: Fuzhou, China, 2017. [Google Scholar]
- Brower, A.V.Z.; DeSalle, R. Patterns of mitochondrial versus nuclear DNA sequence divergence among nymphalid butterflies: The utility of wingless as a source of characters for phylogenetic inference. Insect Mol. Biol. 1998, 7, 73–82. [Google Scholar] [CrossRef]
- Cho, S.W.; Mitchell, A.; Regier, J.C.; Mitter, C.; Poole, R.W.; Friedlander, T.P.; Zhao, S.W. A Highly Conserved Nuclear Gene for Low-Level Phylogenetics—Elongation Factor-1-Alpha Recovers Morphology-Based Tree for Heliothine Moths. Mol. Biol. Evol. 1995, 12, 650–656. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Xia, X.H.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.H. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Xia, X.H. DAMBE6: New tools for microbial genomics, phylogenetics and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Z.M.; Gu, X.S.; Wang, M.; Huang, G.H.; Zwick, A. Phylogenetic relationships among Bombycidae s.l. (Lepidoptera) based on analyses of complete mitochondrial genomes. Syst. Entomol. 2019, 44, 490–498. [Google Scholar] [CrossRef]
- Liu, N.; Li, N.; Yang, P.; Sun, C.; Fang, J.; Wang, S. The complete mitochondrial genome of Damora sagana and phylogenetic analyses of the family Nymphalidae. Genes Genom. 2018, 40, 109–122. [Google Scholar] [CrossRef]
- Tian, L.; Sun, X.-Y.; Chen, M.; Gai, Y.-H.; Hao, J.-S.; Yang, Q. Complete mitochondrial genome of the Five-dot Sergeant Parathyma sulpitia (Nymphalidae: Limenitidinae) and its phylogenetic implications. Zool. Res. 2012, 33, 133–143. [Google Scholar] [CrossRef]
- Wang, X.C.; Sun, X.Y.; Sun, Q.Q.; Zhang, D.X.; Hu, J.; Yang, Q.; Hao, J.S. Complete mitochondrial genome of the laced fritillary Argyreus hyperbius (Lepidoptera: Nymphalidae). Zool. Res. 2011, 32, 465–475. [Google Scholar]
- Kim, M.I.; Baek, J.Y.; Kim, M.J.; Jeong, H.C.; Kim, K.G.; Bae, C.H.; Han, Y.S.; Jin, B.R.; Kim, I. Complete nucleotide sequence and organization of the mitogenome of the red-spotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Mol. Cells 2009, 28, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.S.; Song, L.; Zhou, L.; Shi, Y.X.; Song, N.; Zhang, Y.L. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: Papilionoidea). Int. J. Biol. Macromol. 2020, 145, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNA (serAGN) that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Chen, L.; Wahlberg, N.; Liao, C.Q.; Wang, C.B.; Ma, F.Z.; Huang, G.H. Fourteen complete mitochondrial genomes of butterflies from the genus Lethe (Lepidoptera, Nymphalidae, Satyrinae) with mitogenome-based phylogenetic analysis. Genomics 2020, 112, 4435–4441. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.H. Molecular phylogeny and historical biogeography of the main groups of nymphalid butterflies (Lepidoptera: Papilionoidea: Nymphalidae). Ph.D. Thesis, Anhui Normal University, Wuhu, China, 2015. [Google Scholar]
- Wahlberg, N. That awkward age for butterflies: Insights from the age of the butterfly subfamily Nymphalinae (Lepidoptera: Nymphalidae). Syst. Biol. 2006, 55, 703–714. [Google Scholar] [CrossRef]
- Nylin, S.; Wahlberg, N. Does plasticity drive speciation? Host-plant shifts and diversification in nymphaline butterflies (Lepidoptera: Nymphalidae) during the tertiary. Biol. J. Linn. Soc. 2008, 94, 115–130. [Google Scholar] [CrossRef]
- Leneveu, J.; Chichvarkhin, A.; Wahlberg, N. Varying rates of diversification in the genus Melitaea (Lepidoptera: Nymphalidae) during the past 20 million years. Biol. J. Linn. Soc. 2009, 97, 346–361. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, Y.Z.; Zhou, X.; Kong, X.B.; Wei, S.J.; Ward, R.D.; Zhang, A.B. Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes. BMC Genomics 2015, 16, 428. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Chen, M.Y.; Wang, J.F.; Liang, A.P.; Lin, C.P. Some mitochondrial genes perform better for damselfly phylogenetics: Species- and population-level analyses of four complete mitogenomes of Euphaea sibling species. Syst. Entomol. 2018, 43, 702–715. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Wang, H.; Fang, L.; Zhang, Y. Mitogenome of the Doleschallia bisaltide and Phylogenetic Analysis of Nymphalinae (Lepidoptera, Nymphalidae). Diversity 2023, 15, 558. https://doi.org/10.3390/d15040558
Liu N, Wang H, Fang L, Zhang Y. Mitogenome of the Doleschallia bisaltide and Phylogenetic Analysis of Nymphalinae (Lepidoptera, Nymphalidae). Diversity. 2023; 15(4):558. https://doi.org/10.3390/d15040558
Chicago/Turabian StyleLiu, Ning, Hao Wang, Lijun Fang, and Yalin Zhang. 2023. "Mitogenome of the Doleschallia bisaltide and Phylogenetic Analysis of Nymphalinae (Lepidoptera, Nymphalidae)" Diversity 15, no. 4: 558. https://doi.org/10.3390/d15040558
APA StyleLiu, N., Wang, H., Fang, L., & Zhang, Y. (2023). Mitogenome of the Doleschallia bisaltide and Phylogenetic Analysis of Nymphalinae (Lepidoptera, Nymphalidae). Diversity, 15(4), 558. https://doi.org/10.3390/d15040558