
Unraveling Techniques for Plant Microbiome Structure Analysis

{kind=link}
{kind=link}
Abstract
:1. Introductions: Agriculture, Food production and Problems
2. Microorganisms: Alternatives to Chemical Fertilizers and Pesticides
3. Plant Microbiota
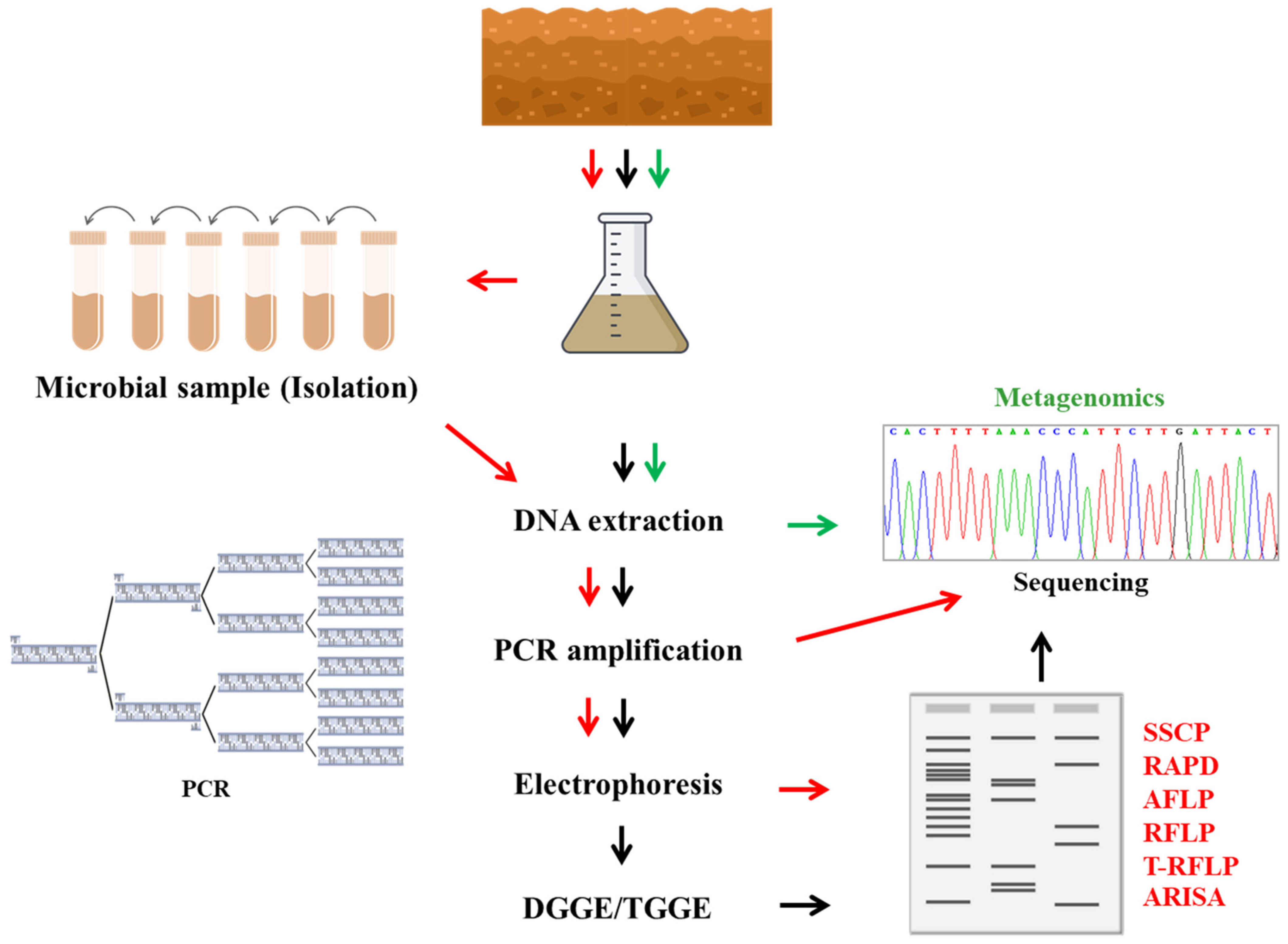
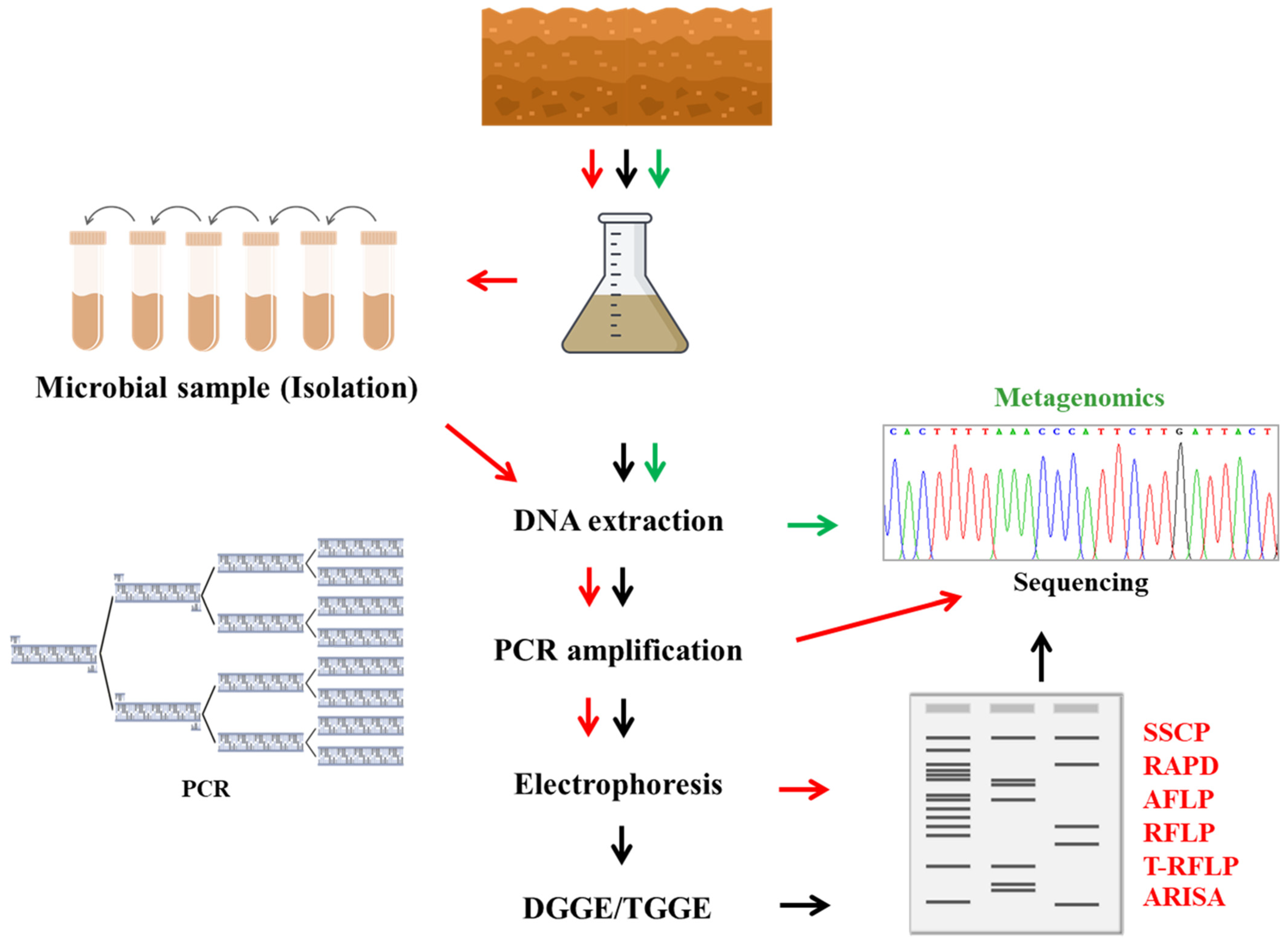
4. The Microbial Identification and Characterization Methods
5. Biochemical Techniques to Determine the Microbial Diversity
6. Genetics Techniques
6.1. Nucleic Acid Hybridization and Fluorescent In Situ Hybridization (FISH)
6.2. Guanine/Cytosine (G + C)
6.3. PCR-Based Techniques
6.3.1. Denaturing Gradient Gel Electrophoresis (DGGE) and Temperature Gradient Gel Electrophoresis (TGGE)
6.3.2. Length Polymorphisms
7. Metagenomics Approach
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Basosi, R.; Spinelli, D.; Fierro, A.; Jez, S. Mineral Nitrogen Fertilizers: Environmental Impact of Production and Use; Fernando Lòpez-Valdez, F.F.L., Ed.; NOVA Science Publishers: Hauppauge, NY, USA, 2014; pp. 3–43. [Google Scholar]
- Ahmed, M.; Rauf, M.; Mukhtar, Z.; Saeed, N.A. Excessive use of nitrogenous fertilizers: An unawareness causing serious threats to environment and human health. Environ. Sci. Pollut. Res. Int. 2017, 24, 26983–26987. [Google Scholar] [CrossRef] [PubMed]
- Hakeem, K.R.; Sabir, M.; Ozturk, M.; Akhtar, M.S.; Ibrahim, F.H. Nitrate and nitrogen oxides: Sources, health effects and their remediation. Rev. Environ. Contam. Toxicol. 2017, 242, 183–217. [Google Scholar] [CrossRef] [PubMed]
- Singh, B. Are nitrogen fertilizers deleterious to soil health? Agronomy 2018, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Chriki, S.; Hocquette, J.-F. The Myth of cultured meat: A review. Front. Nutr. 2020, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.F.; Olivares, F.L. Plant microbiome structure and benefits for sustainable agriculture. Curr. Plant Biol. 2021, 26, 100198. [Google Scholar] [CrossRef]
- Dubey, S.; Sharma, S. Rhizospheric engineering by plant-mediated indirect selection of microbiome for agricultural sustainability. Crit. Rev. Plant Sci. 2021, 40, 379–397. [Google Scholar] [CrossRef]
- Jat, S.L.; Suby, S.B.; Parihar, C.M.; Gambhir, G.; Kumar, N.; Rakshit, S. Microbiome for sustainable agriculture: A review with special reference to the corn production system. Arch. Microbiol. 2021, 203, 2771–2793. [Google Scholar] [CrossRef] [PubMed]
- Kavamura, V.N.; Mendes, R.; Bargaz, A.; Mauchline, T.H. Defining the wheat microbiome: Towards microbiome-facilitated crop production. Comput. Struct. Biotechnol. J. 2021, 19, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Bakker, P.A.; Berendsen, R.L.; Doornbos, R.F.; Wintermans, P.C.; Pieterse, C.M. The rhizosphere revisited: Root microbiomics. Front. Plant Sci. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmanan, V.; Selvaraj, G.; Bais, H.P. Functional soil microbiome: Belowground solutions to an aboveground problem. Plant Physiol. 2014, 166, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, P.; Bhuyan, S.K.; Yadava, P.K.; Varma, A.; Tuteja, N. Emergence of plant and rhizospheric microbiota as stable interactomes. Protoplasma 2017, 254, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Panke-Buisse, K.; Poole, A.C.; Goodrich, J.K.; Ley, R.E.; Kao-Kniffin, J. Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 2015, 9, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S. Disentangling the factors shaping microbiota composition across the plant holobiont. New Phytol. 2016, 209, 454–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacquard, S.; Schadt, C.W. Towards a holistic understanding of the beneficial interactions across the Populus microbiome. New Phytol. 2015, 205, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.G.; Németh, J.B.; Barry, K.; Hainaut, M.; Henrissat, B.; Johnson, J.; Kuo, A.; Lim, J.H.P.; Lipzen, A.; Nolan, M.; et al. Comparative genomics provides insights into the lifestyle and reveals functional heterogeneity of dark septate endophytic fungi. Sci. Rep. 2018, 8, 6321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiza, L.; St-Arnaud, M.; Yergeau, E. Harnessing phytomicrobiome signaling for rhizosphere microbiome engineering. Front. Plant Sci. 2015, 6, 507. [Google Scholar] [CrossRef] [PubMed]
- Solbrig, O.T. The origin and function of biodiversity. Environ. Sci. Policy Sustain. Dev. 1991, 33, 16–38. [Google Scholar] [CrossRef]
- Godfray, H.C.J.; Lawton, J.H. Scale and species numbers. Trends Ecol. Evol. 2001, 16, 400–404. [Google Scholar] [CrossRef]
- Øvreås, L. Population and community level approaches for analysing microbial diversity in natural environments. Ecol. Lett. 2000, 3, 236–251. [Google Scholar] [CrossRef]
- Hughes, J.B.; Hellmann, J.J.; Ricketts, T.H.; Bohannan, B.J. Counting the uncountable: Statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 2001, 67, 4399–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Dash, H.R. Microbial Diversity in the Genomic Era; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Rashid, M.; Stingl, U. Contemporary molecular tools in microbial ecology and their application to advancing biotechnology. Biotechnol. Adv. 2015, 33, 1755–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grządziel, J.; Gałązka, A. The molecular-based methods used for studying bacterial diversity in soils contaminated with PAHs (The Review). In Soil Contamination—Current Consequences and Further Solutions; Marcelo, L., Larramendy, S.S., Eds.; IntechOpen: London, UK, 2016; pp. 85–104. [Google Scholar]
- Elsayed, T.R.; Grosch, R.; Smalla, K. Potato plant spheres and to a lesser extent the soil type influence the proportion and diversity of bacterial isolates with in vitro antagonistic activity towards Ralstonia solanacearum. FEMS Microbiol. Ecol. 2021, 97, fiab038. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Li, J.H.; Qi, Y.H.; Guo, W.; Li, X.; Li, M.Q. Effects of naked barley root rot on rhizosphere soil microorganisms and enzyme activity. Acta Ecol. Sin. 2017, 37, 5640–5649. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, X.; Yan, A. Microbial profiles of retail pacific oysters (Crassostrea gigas) from Guangdong province, China. Front. Microbiol. 2021, 12, 689520. [Google Scholar] [CrossRef] [PubMed]
- Tabacchioni, S.; Chiarini, L.; Bevivino, A.; Cantale, C.; Dalmastri, C. Bias caused by using different isolation media for assessing the genetic diversity of a natural microbial population. Microb. Ecol. 2000, 40, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef]
- van Elsas, J.D.; Duarte, G.F.; Keijzer-Wolters, A.; Smit, E. Analysis of the dynamics of fungal communities in soil via fungal-specific PCR of soil DNA followed by denaturing gradient gel electrophoresis. J. Microbiol. Methods 2000, 43, 133–151. [Google Scholar] [CrossRef]
- Escobar Ortega, J.S.; Aguilar Vásquez, N.N.; Ávila Alba, T.; García de Salamone, I.E. Impact of management of cover crop–soybean agroecosystems on rhizosphere microbial communities. Eur. J. Soil Sci. 2021, 72, 1154–1176. [Google Scholar] [CrossRef]
- Hu, H.; Li, X.; Wu, S.; Lou, W.; Yang, C. Effects of long-term exposure to oxytetracycline on phytoremediation of swine wastewater via duckweed systems. J. Hazard. Mater. 2021, 414, 125508. [Google Scholar] [CrossRef]
- Zuluaga, M.Y.A.; Milani, K.M.L.; Miras-Moreno, M.B.; Lucini, L.; Valentinuzzi, F.; Mimmo, T.; Pii, Y.; Cesco, S.; Rodrigues, E.P.; de Oliveira, A.L.M. The adaptive metabolomic profile and functional activity of tomato rhizosphere are revealed upon PGPB inoculation under saline stress. Environ. Exp. Bot. 2021, 189, 104552. [Google Scholar] [CrossRef]
- Ginting, R.; Solihat, N.; Hafsari, A. Potential bacteria capable of remediating mercury contaminated soils. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Bogor, Indonesia, 16–18 September 2020; IOP Publishing: Bristol, UK, 2020; p. 012136. [Google Scholar]
- Kim, Y.S.; Balaraju, K.; Jeon, Y.H. Biological characteristics of Bacillus amyloliquefaciens AK-0 and suppression of ginseng root rot caused by Cylindrocarpon destructans. J. Appl. Microbiol. 2017, 122, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Yinli, Z.; Jianwei, G.; Wei, Y.; Jianbo, B.; Xianqi, H. Study on isolation, identification and colonization ability of antagonistic bacterium M39 against pomegranate wilt pathogen. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Guiyang, China, 14–16 June 2019; IOP Publishing: Bristol, UK, 2019; p. 052019. [Google Scholar]
- Khatri, K.; Mohite, J.A.; Pandit, P.S.; Bahulikar, R.; Rahalkar, M.C. Description of ‘Ca. Methylobacter oryzae’ KRF1, a novel species from the environmentally important Methylobacter clade 2. Antonie Van Leeuwenhoek 2020, 113, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Nwigwe, C.; Fossey, A.; de Smidt, O. Characterisation of Eucalyptus rhizospheric communities using fatty acid methyl ester (FAME) profile analysis. S. Afr. J. Plant Soil. 2021, 38, 116–125. [Google Scholar] [CrossRef]
- Poblete-Morales, M.; Carvajal, D.; Almasia, R.; Michea, S.; Cantillana, C.; Levican, A.; Silva-Moreno, E. Pseudomonas atacamensis sp. nov., isolated from the rhizosphere of desert bloom plant in the region of Atacama, Chile. Antonie Van Leeuwenhoek 2020, 113, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Park, C.; Lee, B.H.; Lee, K.E.; Park, W. Bacillus miscanthi sp. nov., a alkaliphilic bacterium from the rhizosphere of Miscanthus sacchariflorus. Int. J. Syst. Evol. Microbiol. 2020, 70, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.-D.; Li, Z.; Ping, C.; Qing, D.; Ting, P.; Chun, S.; Wang, X.-C.; Liu, W.-G.; Yang, W.-Y.; Yong, T.-W. Effects of maize-soybean relay intercropping on crop nutrient uptake and soil bacterial community. J. Integr. Agric. 2019, 18, 2006–2018. [Google Scholar] [CrossRef]
- Luo, Y.; Xiao, M.; Yuan, H.; Liang, C.; Zhu, Z.; Xu, J.; Kuzyakov, Y.; Wu, J.; Ge, T.; Tang, C. Rice rhizodeposition promotes the build-up of organic carbon in soil via fungal necromass. Soil Biol. Biochem. 2021, 160, 108345. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Ge, C.; Yao, H. Fungi dominated the incorporation of 13C-CO2 into microbial biomass in tomato rhizosphere soil under different CO2 concentrations. Microorganisms 2021, 9, 2121. [Google Scholar] [CrossRef]
- Liu, J.; Bao, Y.; Zhang, X.; Xu, S.; Qiu, J.; He, J. Rhodobacter kunshanensis sp. nov., a novel bacterium isolated from activated sludge. Curr. Microbiol. 2021, 78, 3791–3797. [Google Scholar] [CrossRef]
- Posada, L.F.; Álvarez, J.C.; Romero-Tabarez, M.; de-Bashan, L.; Villegas-Escobar, V. Enhanced molecular visualization of root colonization and growth promotion by Bacillus subtilis EA-CB0575 in different growth systems. Microbiol. Res. 2018, 217, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Rasul, M.; Yasmin, S.; Zubair, M.; Mahreen, N.; Yousaf, S.; Arif, M.; Sajid, Z.I.; Mirza, M.S. Phosphate solubilizers as antagonists for bacterial leaf blight with improved rice growth in phosphorus deficit soil. Biol. Control 2019, 136, 103997. [Google Scholar] [CrossRef]
- Thurnheer, T.; Gmür, R.; Guggenheim, B. Multiplex FISH analysis of a six-species bacterial biofilm. J. Microbiol. Methods 2004, 56, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Galaviz, C.; Lopez, B.R.; de-Bashan, L.E.; Hirsch, A.M.; Maymon, M.; Bashan, Y. Root growth improvement of mesquite seedlings and bacterial rhizosphere and soil community changes are induced by inoculation with plant growth-promoting bacteria and promote restoration of eroded desert soil. Land Degrad. Dev. 2018, 29, 1453–1466. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, T.; Wang, P.; Tian, H.; Wang, Y.; Cheng, J. Characterization of diazotrophic growth-promoting rhizobacteria isolated from ginger root soil as antagonists against Ralstonia solanacearum. Biotechnol. Biotechnol. Equip. 2018, 32, 1447–1454. [Google Scholar] [CrossRef]
- Leal, C.; Fontaine, F.; Aziz, A.; Egas, C.; Clément, C.; Trotel-Aziz, P. Genome sequence analysis of the beneficial Bacillus subtilis PTA-271 isolated from a Vitis vinifera (cv. Chardonnay) rhizospheric soil: Assets for sustainable biocontrol. Environ. Microbiome 2021, 16, 3. [Google Scholar] [CrossRef]
- Chhetri, G.; Kim, J.; Kim, I.; Kang, M.; So, Y.; Seo, T. Oryzicola mucosus gen. nov., sp. nov., a novel slime producing bacterium belonging to the family Phyllobacteriaceae isolated from the rhizosphere of rice plants. Antonie Van Leeuwenhoek 2021, 114, 1925–1934. [Google Scholar] [CrossRef]
- Fazal, A.; Wen, Z.; Yang, M.; Liao, Y.; Fu, J.; He, C.; Wang, X.; Jie, W.; Ali, F.; Hu, D. Deciphering the rhizobacterial assemblages under the influence of genetically engineered maize carrying mcry genes. Environ. Sci. Pollut. Res. 2021, 28, 60154–60166. [Google Scholar] [CrossRef]
- Samaddar, S.; Schmidt, R.; Tautges, N.E.; Scow, K. Adding alfalfa to an annual crop rotation shifts the composition and functional responses of tomato rhizosphere microbial communities. Appl. Soil Ecol. 2021, 167, 104102. [Google Scholar] [CrossRef]
- Chu, H.; Wang, H.; Zhang, Y.; Li, Z.; Wang, C.; Dai, D.; Tang, M. Inoculation with ectomycorrhizal fungi and dark septate endophytes contributes to the resistance of Pinus spp. to pine wilt disease. Front. Microbiol. 2021, 12, 687304. [Google Scholar] [CrossRef]
- Elhady, A.; Abbasi, S.; Safaie, N.; Heuer, H. Responsiveness of elite cultivars vs. ancestral genotypes of barley to beneficial rhizosphere microbiome, supporting plant defense against root-lesion nematodes. Front. Plant Sci. 2021, 12, 721016. [Google Scholar] [CrossRef] [PubMed]
- Ran, L.; Li, J.; Xing, Y.; Zhang, J.; Zhou, X. Effects of p-Coumaric acid on the structure and abundance of soil Pseudomonas spp. community. Allelopath. J. 2021, 53, 211–218. [Google Scholar] [CrossRef]
- Zhao, X.; Miao, R.; Guo, M.; Zhou, Y. Effects of Fire Phoenix (a genotype mixture of Fesctuca arundinecea L.) and Mycobacterium sp. on the degradation of PAHs and bacterial community in soil. Environ. Sci. Pollut. Res. Int. 2021, 28, 25692–25700. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Guo, Y.; Yu, C.; Zhixian, Z.; Rongli, M.; Deng, W.; Li, Y.; Hu, X. The dynamics in rhizosphere microbial communities under bacterial wilt resistance by mulberry genotypes. Arch. Microbiol. 2021, 203, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Pratiwi, E.; Satwika, T.; Agus, F. Analysis of peat bacterial diversity in oil palm plantations and a logged forest in Jambi, Indonesia, using PCR-DGGE technique. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Bogor, Indonesia, 16–18 September 2020; IOP Publishing: Bristol, UK, 2020; p. 012200. [Google Scholar]
- González-García, S.; Álvarez-Pérez, J.M.; Sáenz de Miera, L.E.; Cobos, R.; Ibañez, A.; Díez-Galán, A.; Garzón-Jimeno, E.; Coque, J.J.R. Developing tools for evaluating inoculation methods of biocontrol Streptomyces sp. strains into grapevine plants. PLoS ONE 2019, 14, e0211225. [Google Scholar] [CrossRef] [Green Version]
- Mohkam, M.; Nezafat, N.; Berenjian, A.; Mobasher, M.A.; Ghasemi, Y. Identification of Bacillus probiotics isolated from soil rhizosphere using 16S rRNA, recA, rpoB gene sequencing and RAPD-PCR. Probiotics Antimicrob. Proteins 2016, 8, 8–18. [Google Scholar] [CrossRef]
- Keyser, C.A.; De Fine Licht, H.H.; Steinwender, B.M.; Meyling, N.V. Diversity within the entomopathogenic fungal species Metarhizium flavoviride associated with agricultural crops in Denmark. BMC Microbiol. 2015, 15, 249. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Stummer, B.E.; Guo, Q.; Zhang, W.; Zhang, X.; Zhang, L.; Harvey, P.R. Quantification of Pseudomonas protegens FD6 and Bacillus subtilis NCD-2 in soil and the wheat rhizosphere and suppression of root pathogenic Rhizoctonia solani AG-8. Biol. Control 2021, 154, 104504. [Google Scholar] [CrossRef]
- Aserse, A.A.; Räsänen, L.A.; Aseffa, F.; Hailemariam, A.; Lindström, K. Diversity of sporadic symbionts and nonsymbiotic endophytic bacteria isolated from nodules of woody, shrub, and food legumes in Ethiopia. Appl. Microbiol. Biotechnol. 2013, 97, 10117–10134. [Google Scholar] [CrossRef]
- Moller, A.M.; Valle, M.C.E.; Teyer, L.F.S.; Rosales, G.V.; Bejar, A.A.G.; Hernandez, M.E.T. Genetic variability in Rhizoctonia solani Isolated from Vitis vinifera based on amplified fragment length polymorphism. Am. J. Agric. Biol. Sci. 2011, 6, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Jacobs-Hoffman, I.; Hills, P.N. Effects of the commercial biostimulant BC204 on the rhizosphere microbial community of Solanum lycopersicum L. S. Afr. J. Bot. 2021, 143, 52–60. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, R.; Zhang, R.; Jiang, W.; Chen, X.; Yin, C.; Mao, Z. Isolation, identification, and antibacterial mechanisms of Bacillus amyloliquefaciens QSB-6 and its effect on plant roots. Front. Microbiol. 2021, 12, 746799. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Monje, D.; Lopez Mejia, J. Botanical microbiomes on the cheap: Inexpensive molecular fingerprinting methods to study plant-associated communities of bacteria and fungi. Appl. Plant Sci. 2020, 8, e11334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madigan, A.P.; Egidi, E.; Bedon, F.; Franks, A.E.; Plummer, K.M. Bacterial and fungal communities are differentially modified by melatonin in agricultural soils under abiotic stress. Front. Microbiol. 2019, 10, 2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likar, M.; Stres, B.; Rusjan, D.; Potisek, M.; Regvar, M. Ecological and conventional viticulture gives rise to distinct fungal and bacterial microbial communities in vineyard soils. Appl. Soil. Ecol. 2017, 113, 86–95. [Google Scholar] [CrossRef]
- Ondreičková, K.; Gubišová, M.; Gubiš, J.; Klčová, L.; Horník, M. Rhizosphere bacterial communities of Arundo donax grown in soil fertilised with sewage sludge and agricultural by-products. Agriculture 2019, 65, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Ondreičková, K.; Žofajová, A.; Piliarová, M.; Gubiš, J.; Hudcovicová, M. Monitoring of rhizosphere bacterial communities in soil with sewage sludge addition using two molecular fingerprinting methods: Do these methods give similar results? Agriculture 2016, 62, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.; Zhang, C.; Mathews, E.R.; Tang, C.; Franks, A.E. Microbial community dynamics in the rhizosphere of a cadmium hyper-accumulator. Sci. Rep. 2016, 6, 36067. [Google Scholar] [CrossRef]
- Abdeljalil, N.O.-B.; Vallance, J.; Gerbore, J.; Yacoub, A.; Daami-Remadi, M.; Rey, P. Combining potential oomycete and bacterial biocontrol agents as a tool to fight tomato Rhizoctonia root rot. Biol. Control 2021, 155, 104521. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Alavi, M.; Cardinale, M.; Müller, H.; Berg, G. Stenotrophomonas rhizophila DSM14405T promotes plant growth probably by altering fungal communities in the rhizosphere. Biol. Fertil. Soils 2012, 48, 947–960. [Google Scholar] [CrossRef]
- Wang, X.-R.; Szmidt, A.E. Molecular markers in population genetics of forest trees. Scand. J. For. Res. 2001, 16, 199–220. [Google Scholar] [CrossRef]
- Vallance, J.; Le Floch, G.; Déniel, F.; Barbier, G.; Lévesque, C.A.; Rey, P. Influence of Pythium oligandrum biocontrol on fungal and oomycete population dynamics in the rhizosphere. Appl. Environ. Microbiol. 2009, 75, 4790–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanhi, J.S.L.; de Souza, R.S.C.; de Araújo, L.M.; Okura, V.K.; Mieczkowski, P.; Imperial, J.; Arruda, P. Multiplex amplicon sequencing for microbe identification in community-based culture collections. Sci. Rep. 2016, 6, 29543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittelmann, S.; Seedorf, H.; Walters, W.A.; Clemente, J.C.; Knight, R.; Gordon, J.I.; Janssen, P.H. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE 2013, 8, e47879. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Navas-Molina Jose, A.; Mercier, C.; Xu Zhenjiang, Z.; Mahé, F.; He, Y.; Zhou, H.-W.; Rognes, T.; Caporaso, J.G.; Knight, R.; et al. Open-Source Sequence Clustering Methods Improve the State Of the Art. mSystems 2016, 1, e00003–e00015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayfach, S.; Pollard, K.S. Toward accurate and quantitative comparative metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parab, S.; Bussolino, F. Multi-omics: Overview, challenges, and applications. In Microbiome-Host Interactions; CRC Press: Boca Raton, FL, USA, 2021; pp. 13–20. [Google Scholar]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Salvato, F.; Hettich, R.L.; Kleiner, M. Five key aspects of metaproteomics as a tool to understand functional interactions in host-associated microbiomes. PLoS Pathog. 2021, 17, e1009245. [Google Scholar] [CrossRef]
- Zhang, X.; Figeys, D. Perspective and guidelines for metaproteomics in microbiome studies. J. Proteome Res. 2019, 18, 2370–2380. [Google Scholar] [CrossRef]
- Levy, A.; Conway, J.M.; Dangl, J.L.; Woyke, T. Elucidating bacterial gene functions in the plant microbiome. Cell Host Microbe 2018, 24, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Raina, T.K.; Kumar, A.; Singh, J.; Prasad, R. Plant microbiome: A reservoir of novel genes and metabolites. Plant Gene 2019, 18, 100177. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sraphet, S.; Javadi, B. Unraveling Techniques for Plant Microbiome Structure Analysis. Diversity 2022, 14, 206. https://doi.org/10.3390/d14030206
Sraphet S, Javadi B. Unraveling Techniques for Plant Microbiome Structure Analysis. Diversity. 2022; 14(3):206. https://doi.org/10.3390/d14030206
Chicago/Turabian StyleSraphet, Supajit, and Bagher Javadi. 2022. "Unraveling Techniques for Plant Microbiome Structure Analysis" Diversity 14, no. 3: 206. https://doi.org/10.3390/d14030206
APA StyleSraphet, S., & Javadi, B. (2022). Unraveling Techniques for Plant Microbiome Structure Analysis. Diversity, 14(3), 206. https://doi.org/10.3390/d14030206