Abstract
Recently, 3-hydroxy-4-pyridinones have been extensively studied as chelating bidentate agents of metal ions for various biomedical applications. This study reports the structural characterization and density functional theory (DFT) analysis of centrosymmetric calcium complex based on 4-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) acetic acid (1). The structure of complex 1 was determined by X-ray crystallography. The 3-hydroxy-4-pyridinone ligand in the studied complex is bound to the calcium ion in the desired monodentate, non-bridging manner. The calcium ion has a coordination number of six and adopts a distorted octahedral geometry. Analyzed geometric characteristics corresponding to hydrogen bonds in the crystal. The theoretical study of intra- and intermolecular interactions utilized DFT with the PBE0-D3/def2-TZVP (Gaussian Inc., Wallingford, CT, USA) level of theory. The charge redistribution in the ligand was studied in comparison with the free acid molecule.
1. Introduction
3-Hydroxy-4-pyridinone (3,4-HP) derivatives represent an excellent family of specific bidentate O,O-donor chelators with a six-membered ring skeleton for various metal ions, which have been an object of great interest in the pharmacological and medicinal chemistry [1,2,3,4,5]. Their metal-chelating abilities have been extensively studied, and in all the cases, as already found for the stability constants, the affinity of the 3,4-HPs towards bioactive metals Zr(IV), Al(III), Ga(III), Fe(III), ln(III), Y(III), La(III), Mg(II), Cu(II), Zn(II), was significantly higher than for Ca(II), with a difference in more than two order of magnitude [6,7]. Notably, the Ca(II) compounds were previously synthesized but lacked X-ray characterization until this study.
2. Results
The complex containing calcium(II) ion was synthesized following the method similar to that described previously, which is based on the reaction of the ligand with a metal ion salt (CaCl2) in an aqueous solution with a significant excess of (3-hydroxy-2-methyl-4-oxo-4H-pyridin-1-yl)acetic acid in an alkaline NaOH solution [8,9]. The colorless crystals were examined by X-ray crystallographic analysis, and the structure presented in Figure 1 was obtained. X-ray analysis revealed that complex 1 is a centrosymmetric compound in which the calcium atom is bound to two monodentate acid residues and four water molecules (Figure 1). Thus, the calcium atom has a coordination number of six. The coordination environment is an octahedron. Water molecules occupy equatorial positions. Since the molecule lies in the center of inversion, the oxygen atoms form a plane in which the central calcium atom is located. The organic ligand in 1 is monodentate, although similar hydroxypyridinones are usually considered as bidentate ligands for other important divalent metals such as Mg(II), Cu(II), Zn(II), Sn(II), and Mn(II) [6,7,10]. This difference correlates well with experimental data showing that the chelating capacity of similar hydroxypyridinones decreases in the following ion series: Fe(III) > Al(III) > Cu(II), Zn(II), Pb(II) > Mg(II), Mn(II), Ca(II). Aluminum and copper showed the greatest competition against iron, while magnesium, calcium, and manganese had little or no effect and the other metals an intermediate effect [1].
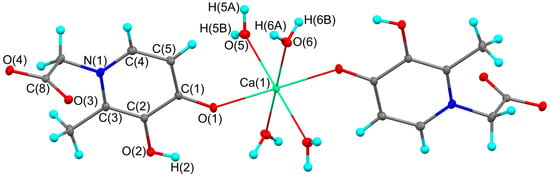
Figure 1.
The molecular structure of complex 1. Thermal ellipsoids are drawn at 30% probability level. Symmetry transformation used to generate equivalent atoms: −x, −y + 1, −z + 1.
The main geometric characteristics in organic fragment of complex 1 are in good agreement with parent acid (Table 1) [11]. As expected, the Ca-O distance between the metal atom and the charged ligand (2.2710(6) Å) is significantly shorter compared to the distances to the coordinated neutral water molecules (2.3542(6) and 2.3694(6) Å). These values are in excellent agreement with previously published related six-coordinate calcium complexes [12,13,14,15,16]. The O(1)…H(2) distance in 1 (2.215(15) Å) is comparable to those in o-catecholates and indicates the possible implementation of an intramolecular O-H…O contact [17]. As with the parent acid [11], the plane of the carboxyl group is almost orthogonal to the main plane of the ligand. The corresponding dihedral angles are 85.13(6) and 89.89(5)° in HL and complex 1, respectively.

Table 1.
The selected bond lengths [Å] and angles [deg] in the complex 1 and 4-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) acetic acid (HL) [11].
The two C-O distances in the carboxyl group of the ligand of complex 1 differ, as in the parent acid [11]. Analysis of the Cambridge Structural Database (CSD) [18] shows that there are many examples of related compounds in which the C-O distances in the deprotonated carboxyl group are largely aligned [19,20,21,22,23,24]. The significant difference in the lengths of the C-O bonds in the carboxyl group in complex 1 (as well as in the parent acid) is probably determined by intermolecular interactions in the crystal. The hydrogen atoms of the coordinated water molecules and the hydroxyl group of the ligand of one molecule form short H…O contacts with the carboxyl group of the neighboring molecule (Table 2).
Table 2.
Geometrical characteristics corresponding to O–H…O short contacts in crystal 1.
As a result, dimer pairs are formed in the crystal (Figure 2). It is interesting to note that the distances between the centers of C-C bonds in the six-membered ligand cycles of neighboring molecules in a dimer pair (3.55 Å) are shorter than the geometric criterion for the existence of a π…π interaction (3.80 Å) [25]. This may indicate a partial overlap of π-systems of ligands and additional intermolecular stacking. Dimer pairs, due to the implementation of hydrogen bonds with their neighboring parts, form more complex 3D networks in the crystal (see Supplementary Materials Figure S1). The five intermolecular O-H…O contacts in the crystal 1 (Table 2) can be classified as so-called contracted interactions (<2.15 Å). Contracted intermolecular contacts usually denote that the presence of specific interactions, which are much stronger than ordinary van der Waals interactions, are attractive in nature and significantly affect the structure and properties of molecular crystals [26].
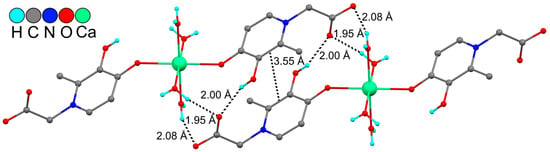
Figure 2.
Fragment of crystal packing of complex 1. Some hydrogen atoms are omitted for clarity.
Coordination to a metal atom can lead to a significant redistribution of electron density (ED) in the organic fragment [10]. In this context, we were interested in comparing the electronic structure of the parent acid HL and complex 1. The corresponding theoretical molecular graphs are shown in Figures S2 and S3. As noted above, all main bond lengths and angles in the organic fragment of complex 1 are in fairly good agreement with the data observed in parent HL. In accordance with this fact, the QTAIM charges on the atoms in these compounds are also close to each other (Table S2). The greatest difference is observed for the O(1) and C(1) atoms. The coordination of the oxygen atom to the calcium atom in complex 1 leads to an increase in the negative charge (−1.28e vs. −1.16e in HL), which in turn leads to an increase in the positive charge on the C(1) atom (0.82e vs. 0.61e in HL). Full geometry optimization without taking into account specific intermolecular contacts leads to the alignment of C-O bonds in the carboxyl group of both HL (1.2418–1.2427 Å) and complex 1 (1.2432–1.2456 Å). In the parent acid it is not possible to localize the critical point (3,−1) corresponding to the O(1)…H(2) contact. Both in the optimized structures and in the crystals the intramolecular O…H distance in HL is somewhat larger compared to that in complex 1 (Table 1 and Table S1). In addition to the O(1)…H(2) contact in structure 1, there is also an interaction between the hydrogen atom of the organic ligand H(5C) and the oxygen atom O(5) of the coordinated water molecule. In turn, the oxygen atom of another symmetrically independent water molecule is involved in the implementation of the intramolecular O…O contact (Figure S3). Analysis of the charge distribution shows that both oxygen atoms are negatively charged (Table S2). In the absence of σ-holes on fluorine or oxygen atoms, the occurrence of δ–…δ– interactions is known to be possible due to the correspondence of the accumulation region of deformation electron density (DED) on one of the atoms to its rarefaction region on the second atom [27,28,29,30]. To test this hypothesis, we constructed selected DED cross-sections in planes corresponding to the supposed O…O contacts. In Figure S4, the accumulation region of deformation ED (blue solid lines) of the O(1) atom corresponds to its rarefaction region (red dashed lines) on the O(6A) atom, as in the previously described fluorine…fluorine and oxygen…oxygen interactions. The energy of O…O interactions in 1 (1.8–2.6 kcal/mol) is comparable to the values for the H(5C)…O(5) contact (1.2 kcal/mol), but is significantly less than the energy of the intraligand hydrogen bond O(1)…H(2) (8.2 kcal/mol). Interaction energies are calculated in accordance with the Espinosa–Molins–Lecomte correlation [31]. It should be noted that the maxima of the DED of oxygen atoms are directed exactly towards the calcium atom. According to Bader’s theory [32,33], Ca-O interactions, as well as non-covalent intramolecular contacts in 1, are closed-shell interactions (laplacian of electron density ∇2ρ(rcp) > 0, local electron energy density he(rcp) > 0). The energy of Ca-O interactions with an organic ligand, as expected, is somewhat higher compared to the energy of Ca-O coordination bonds with water molecules (Table S3).
To estimate the energy of intermolecular interactions within the dimer pair, a full optimization of the geometry of the corresponding associate was carried out at the PCM(H2O)-PBE0-D3/def2-TZVP level (Figure 3). Confirming the existence of stacking between the six-membered rings of ligands of neighboring molecules, a critical point (3,−1) was observed between the C(2) atoms. The energy of the corresponding interaction is 1.0 kcal/mol. It should be noted that the energy of intermolecular hydrogen bonds varies over a very wide range from 1.5 to 18.6 kcal/mol (Table 3). As a result, the calculated total energy of all intermolecular contacts within the dimer pair is 92.4 kcal/mol. In contrast to intramolecular hydrogen bonds, strong intermolecular contacts can be classified as intermediate interactions (∇2ρ(rcp) > 0, he(rcp) < 0) within the framework of Bader’s theory.
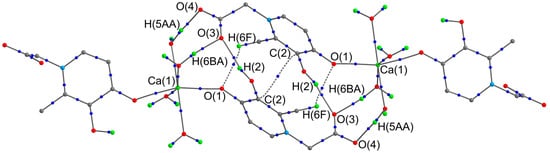
Figure 3.
Theoretical molecular graph of dimeric pair of complex 1. Some hydrogen atoms on organic fragments have been omitted for clarity. The critical points (3,−1) (blue color) of only intermolecular non-covalent interactions are given.
Table 3.
The main topological parameters of ED at (3,−1) critical points corresponding to intermolecular interactions in dimer pair 1.
A Hirshfeld analysis of the intermolecular interactions allows us to detect weaker H…H, H…C, O…C, O…N, C…C, O…O, and H…N contacts (Figure 4 and Figure S5). At the same time, it confirms the largest contribution of O…H contacts (42.8%) to intermolecular interactions in the crystal 1.
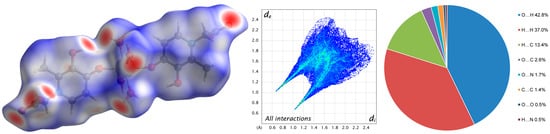
Figure 4.
Molecular Hirshfeld surface (3D d-norm) and 2D fingerprint plot of complex 1.
3. Materials and Methods
3.1. X-Ray Crystallography
The diffraction data for complex 1 were collected with Rigaku OD Xcalibur E diffractometer (Mo-Kα radiation, ω-scan technique, λ = 0.71073 Å). To analyze the obtained data (indexing, data integration, frame scaling, and absorption correction), we used the CrysAlisPro software package (version 1.171.41.93a) [34]. The structure was solved by dual method [35] and refined on using SHELXTL package [36]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms in all water molecules and H(2) atom were located from different Fourier maps and were refined freely without any restrictions. All other hydrogen atoms were placed in calculated positions and were refined in the riding model [Uiso(H) = 1.5Ueq(C) for CH3-group and Uiso(H) = 1.2Ueq(C) for other groups].
Crystal data for 1. C16H24CaN2O12, M = 476.45, monoclinic, space group P21/c, T = 100(2) K, a = 10.1401(2), b = 7.73050(10) and c = 13.2726(2) Å, β = 105.064(2)°, V = 1004.66(3) Å3, Z = 2, dcalc = 1.575 g cm−3, μ = 0.382 mm−1, F000 = 500. A colorless plate-shaped single crystal with dimensions of 0.56 × 0.43 × 0.24 mm was selected, and the intensities of 39,518 reflections were measured using a Rigaku OD Xcalibur E diffractometer (Range of θ: 2.080–34.337°). After merging of equivalents and absorption corrections, 4216 independent reflections (Rint = 0.0369) were used for the structure solution and refinement. Final R factors: R1 = 0.0274 [3740 reflections with I > 2σ(I)], wR2 = 0.0801 (all reflections), GOF = 1.053, Largest diff. peak and hole 0.614 and −0.207e·Å−3.
3.2. Theoretical Methods
The calculations of the non-covalent interactions in crystal 1 were carried out using the Gaussian-09 [37] and the PBE0 [38]-D3 [39]/def2- [40] level of theory. Previously, this level of theory was successfully applied to study non-covalent interactions in the HL crystal and related compounds [6]. Full geometry optimizations were carried out without any symmetry constraints. For all the calculations the polarizable continuum model (PCM) [41] was employed to take into account non-specific solvation effects in a water solution. The lack of imaginary vibration modes for the optimized structures indicated their correspondence to the minima on the potential energy surface. The Bader’s “Atoms in molecules” theory (QTAIM) [32,33] has been used to study the interactions and atomic charges discussed herein by means of the AIMall calculation package version 17.11.14 [42]. The deformation electron density was generated using the Multiwfn v. 3.3.8 program [43] based on the wave function corresponding to the optimized geometry of the complex 1. Hirshfeld surface analysis was conducted using CrystalExplorer (v. 17.5) to elucidate intermolecular interactions [44].
4. Conclusions
A study of the calcium complex based on 4-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) acetic acid was carried out with XRD analysis, DFT modeling, and R.Bader’s analysis of the electron density. The 3-hydroxy-4-pyridinone ligand in a six-coordinate complex Ca(II) is bound to the calcium ion in the desired monodentate, non-bridging manner. When comparing the charge distribution in the organic ligand with the parent acid, the greatest changes were observed for the O(1) and C(1) atoms, which was due to the coordination of this oxygen atom to the metal atom. Intramolecular O…O interactions (1.8–2.6 kcal/mol) in complex 1 are realized due to the correspondence of the accumulation region of deformation electron density (DED) on one of the atoms to its rarefaction region on the second atom. Studies of intermolecular interactions in the crystal were also carried out within the framework of R.Bader’s theory and Hirshfeld surface analysis. Confirming the existence of stacking between the six-membered rings of ligands of neighboring molecules, a critical point (3,−1) was observed between the C(2) atoms (1.0 kcal/mol). The calculated total energy of all intermolecular contacts within the dimer pair is 92.4 kcal/mol. Hirschfeld analysis of intermolecular interactions allows us to detect weaker intermolecular contacts but confirms the largest contribution of O…H contacts to the energy of intermolecular interactions (42.8%).
Supplementary Materials
The following supporting information can be downloaded online: Figure S1. Fragment of crystal packing of complex 1. Views along crystallographic a (a), b (b), and c (c) axes; Figure S2. Theoretical molecular graph of HL. Only the critical points (3,−1) (blue color) are given; Figure S3. Theoretical molecular graph of complex 1. Only the critical points (3,−1) (blue color) are given; Table S1. Crystal (X-ray) and optimized (DFT) geometry (the selected bond lengths [Å] and angles [deg]) of complex 1 and 4-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) acetic acid (HL) [S1]. Two values in the DFT column refer to dimensions in the two halves of the molecule; Table S2. Atomic QTAIM charges [e] in the complex 1 and 4-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) acetic acid (HL); Figure S4. Deformation ED distribution in the plane of the intramolecular O(1)…O(6A) interaction. Blue solid lines correspond to the deformation ED concentration region and red dashed lines show its rarefaction region; Table S3. The main topological parameters of ED at selected (3,−1) critical points of complex 1; Figure S5. Molecular Hirshfeld surfaces (3D d-norm) and 2D fingerprint plots of complex 1.
Author Contributions
Conceptualization, M.A.K.; methodology, R.V.R. and G.S.Z.; software, R.V.R.; validation, G.K.F. and S.Y.K.; investigation, R.V.R. and G.S.Z.; resources, M.A.K. and S.Y.K.; writing—original draft preparation, R.V.R. and M.A.K.; writing—review and editing, G.K.F. and S.Y.K.; visualization, R.V.R.; supervision, S.Y.K.; project administration, M.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Data Availability Statement
CCDC 2492822 contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures (accessed on 23 October 2025).
Acknowledgments
This work was supported by the Ministry of Science and Higher Education of the Russian Federation within the scientific tasks of the Razuvaev Institute of Organometallic Chemistry RAS.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Santos, M.A.; Irto, A.; Buglyo, P.; Chaves, S. Hydroxypyridinone-based metal chelators towards ecotoxicity: Remediation and biological mechanisms. Molecules 2022, 27, 1966. [Google Scholar] [CrossRef]
- Santos, M.A.; Marques, S.M.; Chaves, S. Hydroxypyridinones as “privileged” chelating structures for the design of medicinal drugs. Coord. Chem. Rev. 2012, 256, 240–259. [Google Scholar] [CrossRef]
- Irto, A.; Cardiano, P.; Chand, K.; Cigala, R.M.; Crea, F.; De Stefano, C.; Gattuso, G.; Sammartano, S.; Santos, M.A. Complexation of environmentally and biologically relevant metals with bifunctional 3-hydroxy-4-pyridinones. J. Mol. Liq. 2020, 319, 114349. [Google Scholar] [CrossRef]
- Katkova, M.A.; Zabrodina, G.S.; Rumyantcev, R.V.; Zhigulin, G.Y.; Skabitsky, I.V.; Fomina, I.G.; Bekker, O.B.; Ketkov, S.Y.; Eremenko, I.L. Insight into design of 3-hydroxy-4-pyridinone functionalized with isoniazid fragment: Structural characterization and antimycobacterial evaluation. Mendeleev Commun. 2024, 34, 850–853. [Google Scholar] [CrossRef]
- Katkova, M.A.; Zhigulin, G.Y.; Baranov, E.V.; Zabrodina, G.S.; Muravyeva, M.S.; Ketkov, S.Y.; Fomina, I.G.; Eremenko, I.L. Specific Features of Binding Bioactive Organic Molecules with the Metallic Matrix of Heteronuclear 3d-4f Structures Containing Soft and Hard Metallocenters Using the Nd(III)–Cu(II) Complex as an Example. Rus. J. Coord. Chem. 2023, 49, 601–611. [Google Scholar] [CrossRef]
- Garcia-Raso, A.; Terron, A.; Fiol, J.J.; Lopez-Zafra, A.; Herreros, M.; Capilla, I.; Dominguez, M.A.; Barcelo-Oliver, M.; Spingler, B.; Frontera, A. Synthesis, X-ray characterization, and DFT study of six deferiprone analogues. J. Mol. Struct. 2025, 1326, 141123. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Abbate, V.; Chen, Y.L.; Ma, Y.; Zhou, T.; Hider, R.C. Hydroxypyridinone Journey into Metal Chelation. Chem. Rev. 2018, 118, 7657–7701. [Google Scholar] [CrossRef] [PubMed]
- Queiros, C.; Amorim, M.J.; Leite, A.; Ferreira, M.; Gameiro, P.; de Castro, B.; Biernacki, K.; Magalhaes, A.; Burgess, J.; Rangel, M. Nickel(II) and Cobalt(II) 3-Hydroxy-4-pyridinone complexes: Synthesis, characterization and speciation studies in aqueous solution. Eur. J. Inorg. Chem. 2011, 2011, 131–140. [Google Scholar] [CrossRef]
- Rangel, M.; Leite, A.; Silva, A.M.N.; Moniz, T.; Nunes, A.; Amorim, M.J.; Queirós, C.; Cunha-Silva, L.; Gameiro, P.; Burgess, J. Distinctive EPR signals provide an understanding of the affinity of bis-(3-hydroxy-4-pyridinonato) copper(II) complexes for hydrophobic environments. Dalton Trans. 2014, 43, 9722–9731. [Google Scholar] [CrossRef]
- Katkova, M.A.; Rumyantcev, R.V.; Zabrodina, G.S.; Fomina, I.G.; Bekker, O.B.; Ketkov, S.Y.; Fukin, G.K.; Eremenko, I.L. Coordination Polymer Based on Pentacopper Metallamacrocyclic Units Featured by 3-Hydroxy-4-pyridinone Ligands: Synthesis, Structure and Biological Activity. Rus. J. Coord. Chem. 2025, 51, 438–446. [Google Scholar] [CrossRef]
- Orvig, C.; Rettig, S.J.; Zhang, Z. 1-Carboxymethyl-3-hydroxy-2-methyl-4(1H)-pyridinone (monoclinic form 2). Acta Crystallogr. Sect. C Struct. Chem. 1994, 50, 1511–1514. [Google Scholar] [CrossRef]
- Salido, M.L.G.; Mascaros, P.A.; Garzon, R.L.; Valero, M.D.G.; Low, J.N.; Gallagher, J.F.; Glidewell, C. Hydrated metal(II) complexes of N-(6-amino-3,4-dihydro-3-methyl-5-nitroso-4-oxopyrimidin-2-yl) derivatives of glycine, glycylglycine, threonine, serine, valine and methionine: A monomeric complex and coordination polymers in one, two and three dimensions linked by hydrogen bonding. Acta Cryst. Sect. B Struct. Sci. 2004, 60, 46–64. [Google Scholar] [CrossRef]
- Perrin, C.L.; Lau, J.S.; Kim, Y.-J.; Karri, P.; Moore, C.; Rheingold, A.L. Asymmetry of the “Strongest” OHO Hydrogen Bond, in the Monoanion of (±)-α,α′-Di-tert-butylsuccinate. J. Am. Chem. Soc. 2009, 131, 13548–13554. [Google Scholar] [CrossRef]
- Hao, X.-M.; Zhao, S.; Wang, H.; Wu, Y.-B.; Yang, D.; Zhang, X.-F.; Xu, Z.-L. In vitro release of theophylline and cytotoxicity of two new metal–drug complexes. Polyhedron 2018, 142, 38–42. [Google Scholar] [CrossRef]
- Elgemeie, G.H.; Fathy, N.M.; Shaarawi, S.; Jones, P.G. trans-Tetraaquabis{(E)-2-cyano-1-[(ethoxycarbonyl)methylsulfanyl]-2-(1-naphthylaminocarbonyl)ethene-1-thiolato}calcium(II) diethyl ether disolvate. Acta Cryst. Sect. E Struct. Rep. Online 2010, 66, m554–m555. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.-D.; Zhang, Z.-H.; Tong, Y.-Y.; Liu, X.-M.; Ma, X.-F.; Xuan, X.-P. Structural Transformation Pathways of Alkaline Earth Family Coordination Polymers Containing 3,3′,5,5′-Biphenyl Tetracarboxylic Acid. Chem. Asian J. 2019, 14, 1970–1976. [Google Scholar] [CrossRef]
- Arsenyev, M.; Baranov, E.; Chesnokov, S.; Abakumov, G. 4,6-Di-tert-butyl-2,3-dihydroxybenzaldehyde. Acta Cryst. Sect. E Struct. Rep. Online 2013, 69, o1565. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mat. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Gao, S.; Huo, L.-H.; Zhao, J.-G. Tetraaquabis[(4-oxo-4H-pyridin-1-yl)acetato]manganese(II). Acta Cryst. Sect. E Struct. Rep. Online 2007, 63, m1514. [Google Scholar] [CrossRef]
- Huang, Y.-Q.; Chen, H.-Y.; Li, Z.-G.; Wang, Q.; Wang, Y.; Cao, X.-Q.; Zhao, Y. Influence of N-donor ancillary ligands on the structures of three cadmium(II) complexes with L-shaped carboxylate ligand. Inorg. Chim. Acta 2017, 466, 71–77. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Gao, S.; Huo, L.-H.; Zhao, H.; Zhao, J.-G.; Ng, S.W. Hexaaquanickel(II) bis[(4-oxo-4H-pyridin-1-yl)acetate] dehydrate. Acta Cryst. Sect. E Struct. Rep. Online 2004, 60, m544. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Gao, S.; Huo, L.-H.; Zhao, J.-G. 3-Hydroxypyridinium-1-acetate monohydrate. Acta Cryst. Sect. E Struct. Rep. Online 2005, 61, o3554–o3555. [Google Scholar] [CrossRef]
- Zhao, H.; Huo, L.-H.; Gao, S.; Zhang, Z.-Y.; Zhao, J.-G.; Ng, S.W. 1-Carboxymethyl-3-hydroxypyridinium chloride-3-hydroxypyridinium-1-acetate (1/1). Acta Cryst. Sect. E Struct. Rep. Online 2004, 60, o1501–o1503. [Google Scholar] [CrossRef]
- Liu, J.-J.; Liu, N.; Lu, Y.-W.; Zhao, G.-Z. Three Photochromic Co-crystals Based on Viologen Moiety. Chin. J. Inorg. Chem. 2021, 37, 937–944. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Zefirov, Y.V.; Zorkii, P.M. New applications of van der Waals radii in chemistry. Russ. Chem. Rev. 1995, 64, 415–428. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kubsik, M.; Kamiński, R.; Woźniak, K.; Dominiak, P.M. From a single molecule to molecular crystal architectures: Structural and energetic studies of selected uracil derivatives. Cryst. Growth Des. 2012, 12, 2508–2524. [Google Scholar] [CrossRef]
- Rumyantsev, R.V.; Fukin, G.K. Intramolecular nonvalent interactions in the EuII2EuIII(μ-ORF)2(μ2-ORF)3(μ3-ORF)2(DME)2 complex. Russ. J. Coord. Chem. 2019, 45, 767–775. [Google Scholar] [CrossRef]
- Rumyantsev, R.V.; Fukin, G.K.; Baranov, E.V.; Cherkasov, A.V.; Kozlova, E.A. Application of the molecular invariom model for the study of interactions involving fluorine atoms in the {YbII2(μ2-OCH(CF3)2)3(μ3-OCH(CF3)2)2YbIII(OCH(CF3)2)2(THF)(Et2O)} Complex. Russ. J. Coord. Chem. 2021, 47, 235–243. [Google Scholar] [CrossRef]
- Rumyantsev, R.V.; Zabrodina, G.S.; Katkova, M.A.; Ketkov, S.Y.; Fukin, G.K. Study of Intramolecular Interactions in the Polyoxovanadate Cluster [(SO4) ⊂ V16O42]6–. J. Struct. Chem. 2023, 64, 1305–1313. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis Pro Software System, version 1.171.41.93a; Rigaku Corporation: Wroclaw, Poland, 2020.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll, Version 17.11.14; TK Gristmill Software: Overland Park, KS, USA, 2013.
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).