
3′H-Spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
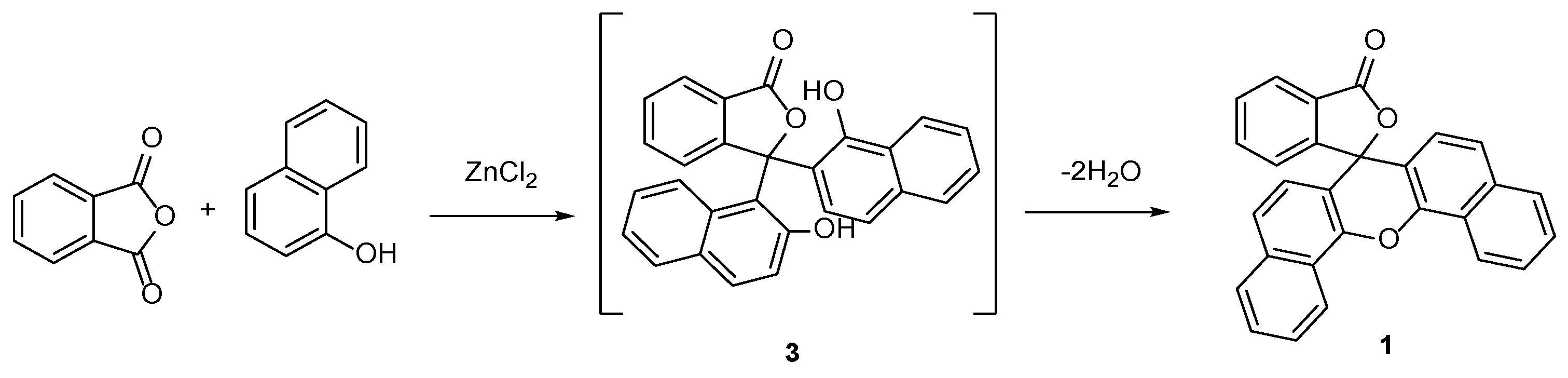
2. Results and Discussion
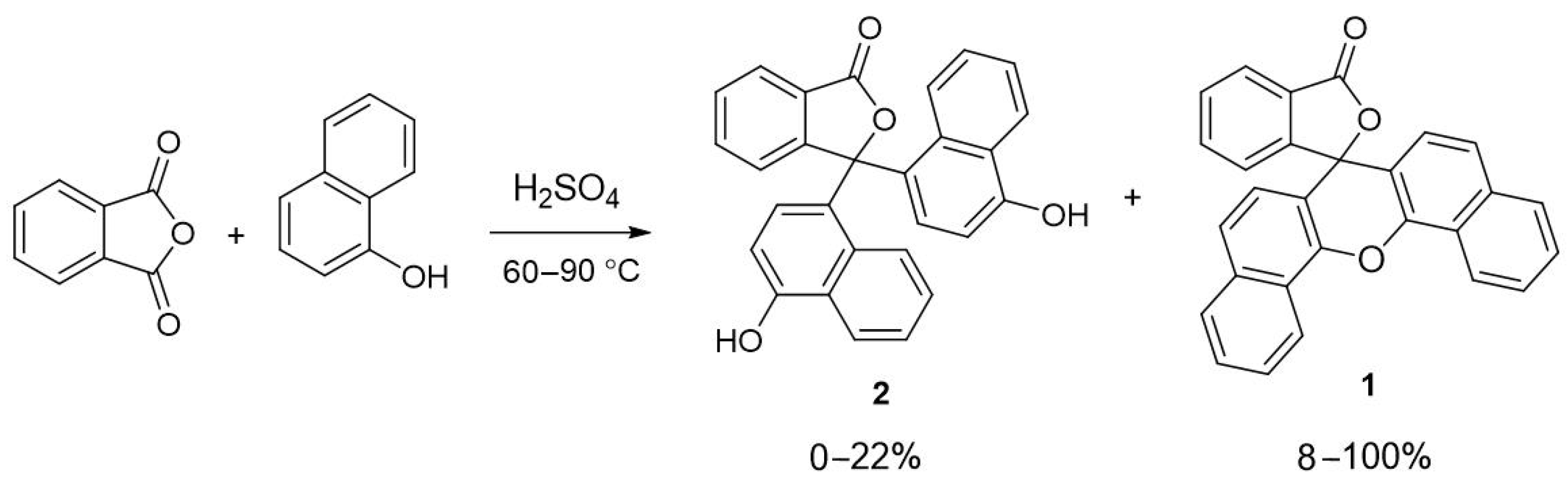
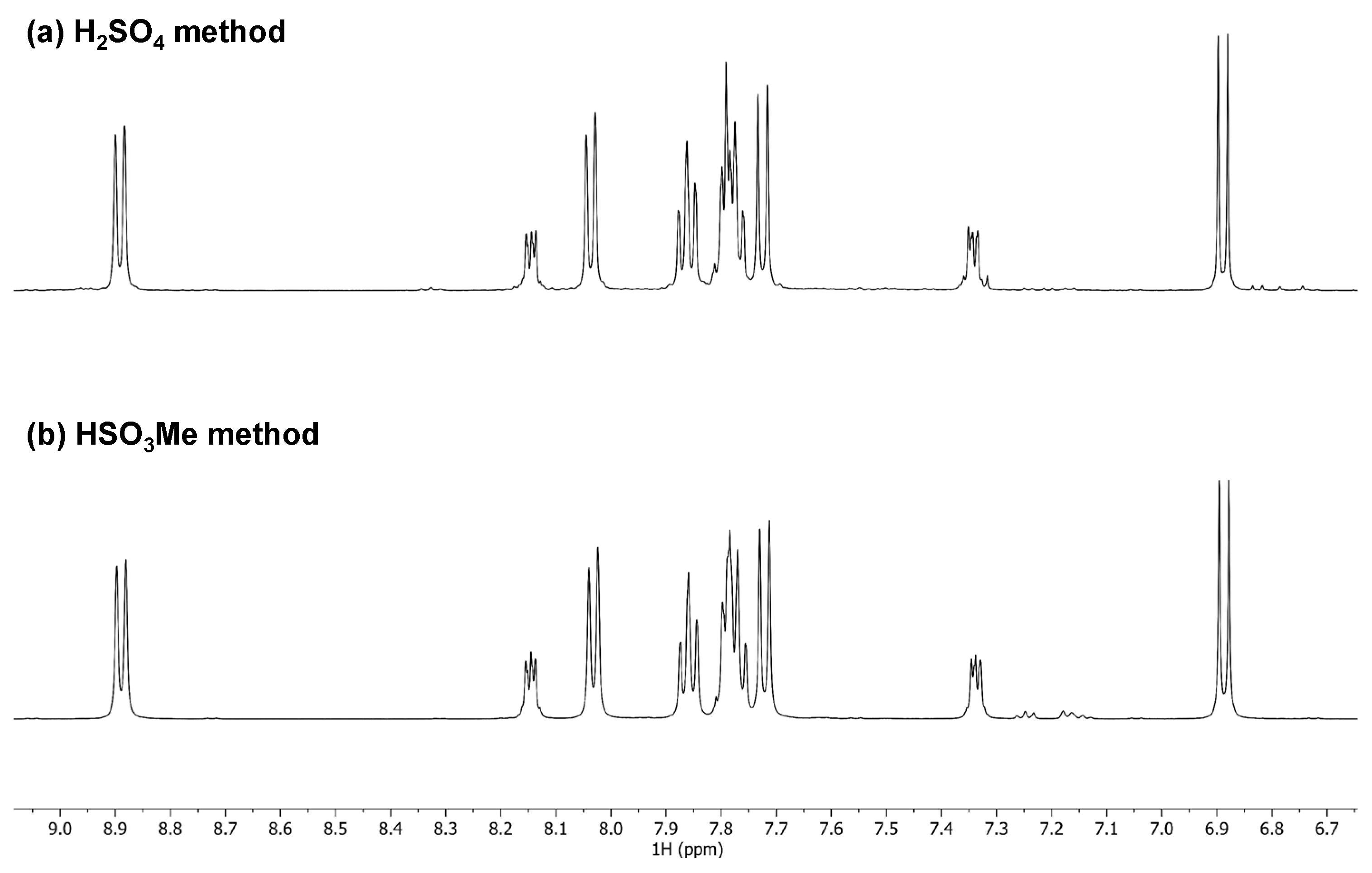
2.1. Synthesis and Spectroscopy
2.2. The Crystal Structure of 1
3. Experimental Section
3.1. 3′H-spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one (1)
3.1.1. Method 1—Methanesulfonic Acid
3.1.2. Method 2—95% Sulfuric Acid
3.2. X-Ray Structure Determination of 1
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabnis, R.W. A facile synthesis of phthalein dyes. Tetrahedron Lett. 2009, 50, 6261–6263. [Google Scholar] [CrossRef]
- Sabnis, R.W. Developments in the chemistry and applications of phthalein dyes. Part 1: Industrial applications. Color. Technol. 2018, 134, 187–205. [Google Scholar] [CrossRef]
- Werner, E.A. III-The Preparation of α-Naphtholphthalein. J. Chem. Soc. Trans. 1918, 113, 20–21. [Google Scholar] [CrossRef]
- Copisarow, M. XXVI-Phthaleins and Fluorans. J. Chem. Soc. Trans. 1920, 117, 209–218. [Google Scholar] [CrossRef]
- Baeyer, A. Ueber die Phenolfarbstoffe. Ber. Dtsch. Chem. Ges. 1871, 4, 658–665. [Google Scholar] [CrossRef]
- Copisarow, M.; Weizmann, C. XCVII-Phthalides of the Benzene, Naphthalene and Carbazole Series. Part 1. J. Chem. Soc. Trans. 1915, 107, 878–886. [Google Scholar] [CrossRef]
- Jones, R.N.; Gallagher, B.S. The infrared spectra of steroid lactones. J. Am. Chem. Soc. 1959, 81, 5242–5251. [Google Scholar] [CrossRef]
- Xu, X.; Strongin, R.M.; Fronczek, R.F. CCDC 1404764; CSD Communication: Omagh, UK, 2015. [Google Scholar] [CrossRef]
- CrysAlisPro v1.171.42.94a & 109a; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2024.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalmers, B.A.; Cordes, D.B.; McKay, A.P.; Patterson, I.L.J.; Vladymyrova, N.; Smellie, I.A. 3′H-Spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one. Molbank 2025, 2025, M2033. https://doi.org/10.3390/M2033
Chalmers BA, Cordes DB, McKay AP, Patterson ILJ, Vladymyrova N, Smellie IA. 3′H-Spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one. Molbank. 2025; 2025(3):M2033. https://doi.org/10.3390/M2033
Chicago/Turabian StyleChalmers, Brian A., David B. Cordes, Aidan P. McKay, Iain L. J. Patterson, Nadiia Vladymyrova, and Iain A. Smellie. 2025. "3′H-Spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one" Molbank 2025, no. 3: M2033. https://doi.org/10.3390/M2033
APA StyleChalmers, B. A., Cordes, D. B., McKay, A. P., Patterson, I. L. J., Vladymyrova, N., & Smellie, I. A. (2025). 3′H-Spiro[dibenzo[c,h]xanthene-7,1′-isobenzofuran]-3′-one. Molbank, 2025(3), M2033. https://doi.org/10.3390/M2033