
[Palladium-decabismuth(4+)]-tetrakis(tetrachloridoaluminate) Cluster Compound, [Pd@Bi10][AlCl4]4: Synthesis, Crystal Structure, and Electronic Structure


Abstract
1. Introduction
2. Results and Discussion
2.1. Crystal Structure
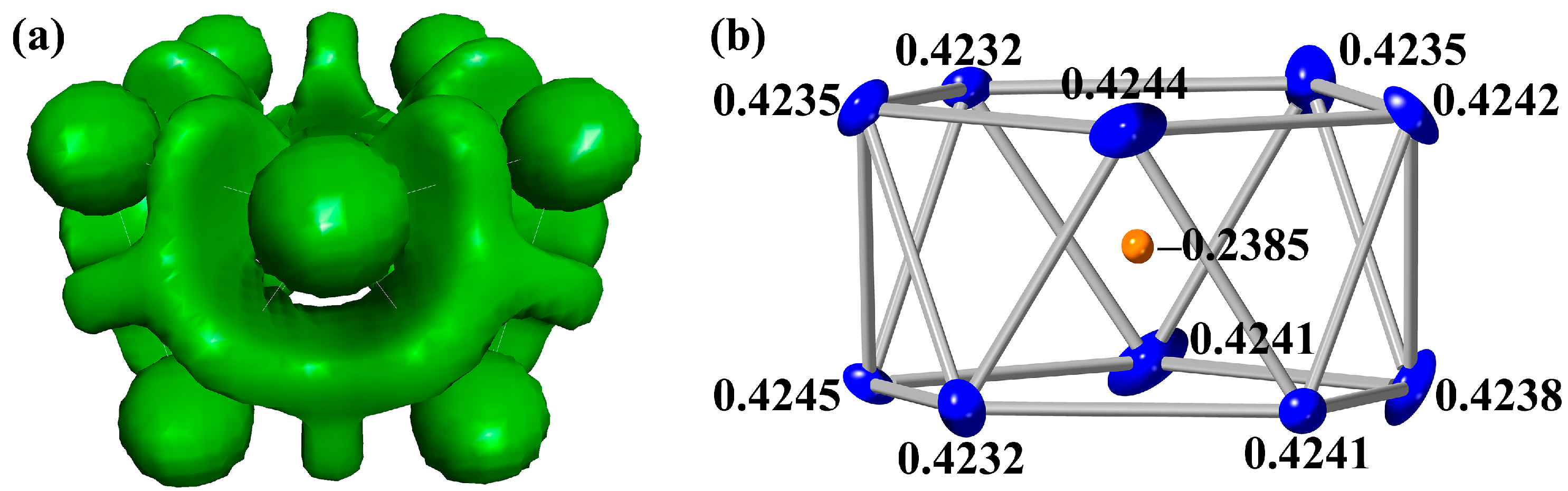
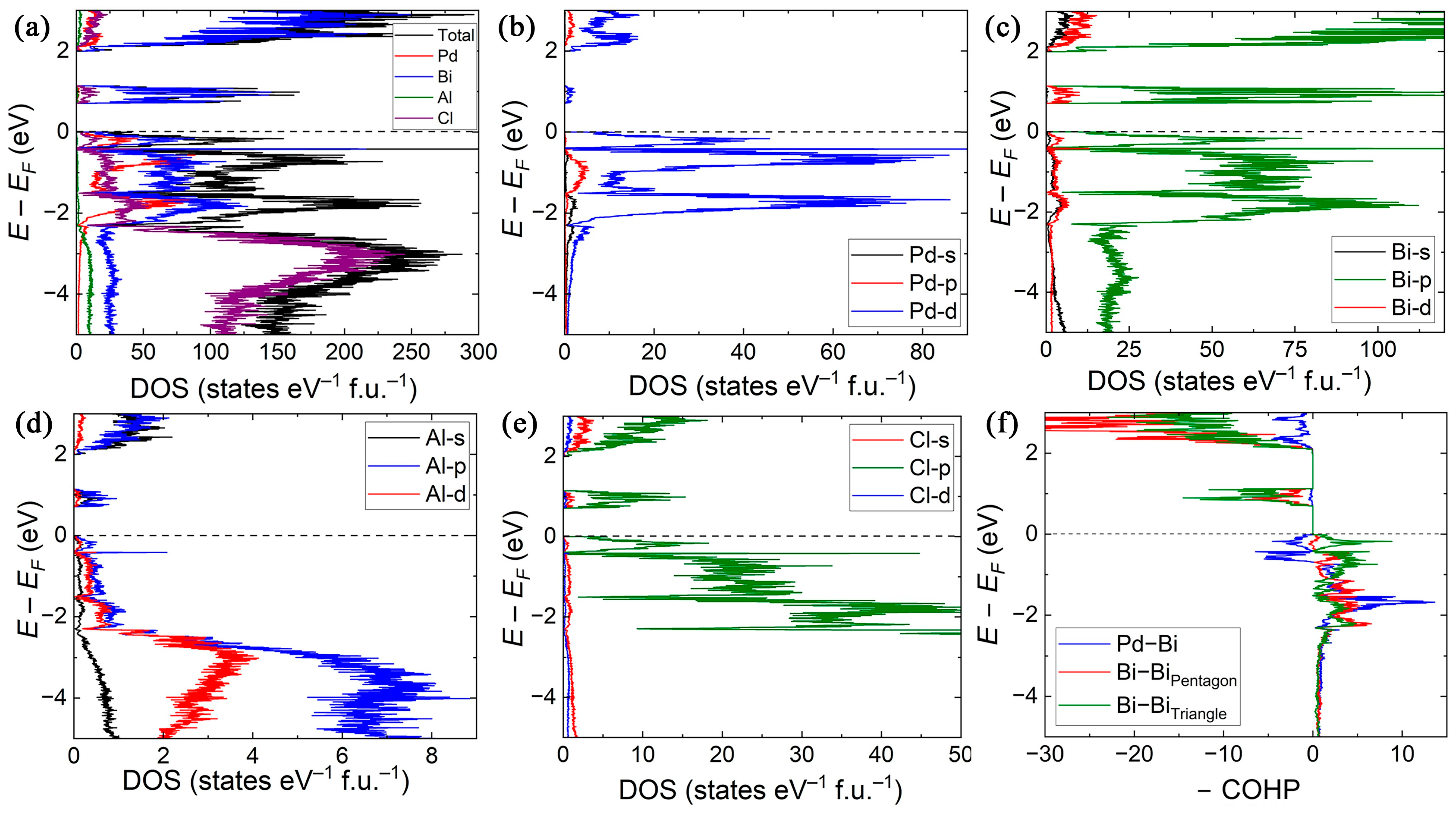
2.2. Electronic Structure
3. Materials and Methods
3.1. Synthesis
3.2. Single Crystal X-Ray Diffraction
3.3. Electronic Structure Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kore, R.; Kelley, S.P.; Sawant, A.D.; Mishra, M.K.; Rogers, R.D. Are ionic liquids and liquid coordination complexes really different?—Synthesis, characterization, and catalytic activity of AlCl3/base catalysts. Chem. Commun. 2020, 56, 5362–5365. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Baaqel, H.; Matthews, R.P.; Chen, Y.; Lovelock, K.R.J.; Hallett, J.P.; Licence, P. Halometallate ionic liquids: Thermal properties, decomposition pathways, and life cycle considerations. Green Chem. 2022, 24, 5800–5812. [Google Scholar] [CrossRef]
- Freudenmann, D.; Wolf, S.; Wolff, M.; Feldmann, C. Ionic liquids: New perspectives for inorganic synthesis? Angew. Chem. Int. Ed. 2011, 50, 11050–11060. [Google Scholar] [CrossRef]
- Ma, Z.; Yu, J.; Dai, S. Preparation of Inorganic Materials Using Ionic Liquids. Adv. Mater. 2010, 22, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Breternitz, J.; Groh, M.F.; Ruck, M. Ionic liquids as crystallization media for inorganic materials. CrystEngComm. 2012, 14, 4874–4885. [Google Scholar] [CrossRef]
- Dupont, J.; Leal, B.C.; Lozano, P.; Monteiro, A.L.; Migowski, P.; Scholten, J.D. Ionic Liquids in Metal, Photo-, Electro-, and (Bio) Catalysis. Chem. Rev. 2024, 124, 5227–5420. [Google Scholar] [CrossRef]
- Zhou, T.; Gui, C.; Sun, L.; Hu, Y.; Lyu, H.; Wang, Z.; Song, Z.; Yu, G. Energy Applications of Ionic Liquids: Recent Developments and Future Prospects. Chem. Rev. 2023, 123, 12170–12253. [Google Scholar] [CrossRef]
- Lei, Z.; Chen, B.; Koo, Y.M.; MacFarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef]
- Baca, K.R.; Al-Barghouti, K.; Wang, N.; Bennett, M.G.; Matamoros Valenciano, L.; May, T.L.; Xu, I.V.; Cordry, M.; Haggard, D.M.; Haas, A.G.; et al. Ionic Liquids for the Separation of Fluorocarbon Refrigerant Mixtures. Chem. Rev. 2024, 124, 5167–5226. [Google Scholar] [CrossRef]
- Lei, Z.; Dai, C.; Hallett, J.; Shiflett, M. Introduction: Ionic Liquids for Diverse Applications. Chem. Rev. 2024, 124, 7533–7535. [Google Scholar] [CrossRef]
- Pan, F.; Peerless, B.; Dehnen, S. Bismuth-Based Metal Clusters horizontal line From Molecular Aesthetics to Contemporary Materials Science. Acc. Chem. Res. 2023, 56, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Köhler, D.; Ruck, M. Room-Temperature Synthesis of Bismuth Clusters in Ionic Liquids and Crystal Growth of Bi5(AlCl4)3. Z. Anorg. Allg. Chem. 2009, 635, 297–300. [Google Scholar] [CrossRef]
- Lindsjö, A.F.M.; Kloo, L. Improvements of and Insights into the Isolation of Bismuth Polycations from Benzene Solution—Single-Crystal Structure Determinations of Bi8[GaCl4]2 and Bi5[GaCl4]3. Eur. J. Inorg. Chem. 2005, 2005, 670–675. [Google Scholar] [CrossRef]
- Krebs, B.; Hucke, M.; Brendel, C.J. Structure of the Octabismuth(2+) Cluster in Crystalline Bi8(AlCl4)2. Angew. Chem. Int. Ed. 1982, 21, 445–446. [Google Scholar] [CrossRef]
- Knies, M.; Ruck, M. Bismuth-rich bimetallic clusters (CuBi8)3+ and [MBi10]4+ (M = Pd, Pt) from ionothermal synthesis. Z. Naturforsch. B, 2022; 77, 191–196. [Google Scholar]
- Groh, M.F.; Wolff, A.; Wahl, B.; Rasche, B.; Gebauer, P.; Ruck, M. Pentagonal Bismuth Antiprisms with Endohedral Palladium or Platinum Atoms by Low-Temperature Syntheses. Z. Anorg. Allg. Chem. 2016, 643, 69–80. [Google Scholar] [CrossRef]
- Groh, M.F.; Müller, U.; Isaeva, A.; Ruck, M. The Intermetalloid Clusters [Ni2Bi12]4+ and [Rh2Bi12]4+– Ionothermal Synthesis, Crystal Structures, and Chemical Bonding. Z. Anorg. Allg. Chem. 2018, 645, 161–169. [Google Scholar] [CrossRef]
- Groh, M.F.; Isaeva, A.; Ruck, M. [Ru2Bi14Br4](AlCl4)4 by mobilization and reorganization of complex clusters in ionic liquids. Chem. Eur. J. 2012, 18, 10886–10891. [Google Scholar] [CrossRef]
- Wahl, B.; Erbe, M.; Gerisch, A.; Kloo, L.; Ruck, M. Nobel-Metal Centered Polycations [Au@Bi10]5+ or [Pd@Bi10]4+ Embedded in Halogenido-Bismuthate(III)-Stannate(II) Frameworks. Z. Anorg. Allg. Chem. 2009, 635, 743–752. [Google Scholar] [CrossRef]
- Knies, M.; Kaiser, M.; Lê Anh, M.; Efimova, A.; Doert, T.; Ruck, M. Low-Temperature Ordering in the Cluster Compound (Bi8)Tl[AlCl4]3. Inorganics 2019, 7, 45. [Google Scholar] [CrossRef]
- Ahmed, E.; Ahrens, E.; Heise, M.; Ruck, M. A Facile Route for the Synthesis of Polycationic Tellurium Cluster Compounds: Synthesis in Ionic Liquid Media and Characterization by Single-Crystal X-Ray Crystallography and Magnetic Susceptibility. Z. Anorg. Allg. Chem. 2010, 636, 2602. [Google Scholar] [CrossRef]
- Ahmed, E.; Breternitz, J.; Groh, M.F.; Isaeva, A.; Ruck, M. [Sb7Se8Br2]3+ and [Sb13Se16Br2]5+—Double and Quadruple Spiro Cubanes from Ionic Liquids. Eur. J. Inorg. Chem. 2014, 19, 3037–3042. [Google Scholar] [CrossRef]
- Müller, U.; Isaeva, A.; Richter, J.; Knies, M.; Ruck, M. Polyhedral Bismuth Polycations Coordinating Gold(I) with Varied Hapticity in a Homoleptic Heavy-Metal Cluster. Eur. J. Inorg. Chem. 2016, 2016, 3580–3584. [Google Scholar] [CrossRef]
- Dubenskyy, V.; Ruck, M. Das Subchlorid Bi16PdCl22: [Pd@Bi10]4+-Polykationen in einem Raumnetzwerk aus Chlorobismutat(III)-Anionen. Z. Anorg. Allg. Chem. 2004, 630, 2458–2462. [Google Scholar] [CrossRef]
- Ruck, M.; Dubenskyy, V.; Sohnel, T. Structure and bonding of Pd@[Bi10]4+ in the subbromide Bi14PdBr16. Angew. Chem. Int. Ed. 2003, 42, 2978–2982. [Google Scholar] [CrossRef]
- Wilson, R.J.; Lichtenberger, N.; Weinert, B.; Dehnen, S. Intermetalloid and Heterometallic Clusters Combining p-Block (Semi)Metals with d- or f-Block Metals. Chem. Rev. 2019, 119, 8506–8554. [Google Scholar] [CrossRef]
- Wahl, B.; Ruck, M. Die molekularen Cluster [Bi10Au2](EBi3X9)2 (E = As, Bi; X = Cl, Br)—Synthese, Kristallstrukturen, Drillingsbildung und chemische Bindung. Z. Anorg. Allg. Chem. 2008, 634, 2267–2275. [Google Scholar] [CrossRef]
- King, R.B.; Silaghi-Dumitrescu, I.; Uţǎ, M.M. Beyond the Wade−Mingos Rules in Bare 10- and 12-Vertex Germanium Clusters: Transition States for Symmetry Breaking Processes. J. Chem. Theory Comput. 2008, 4, 209–215. [Google Scholar] [CrossRef]
- Michael, D.; Mingos, P. Polyhedral skeletal electron pair approach, Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Baranets, S.; Bobev, S. Ca14AlBi11—A new Zintl phase from earth-abundant elements with a great potential for thermoelectric energy conversion. Mater. Today Adv. 2020, 7, 100094. [Google Scholar] [CrossRef]
- Janzen, R.; Baranets, S.; Bobev, S. Synthesis and structural characterization of the new Zintl phases Eu10Mn6Bi12 and Yb10Zn6Sb12. Dalton Trans. 2022, 51, 13470–13478. [Google Scholar] [CrossRef]
- Mednikov, E.G.; Eremenko, N.K.; Mikhailov, V.A.; Gubin, S.P.; Slovokhotov, Y.L.; Struchkov, Y.T. New palladium cluster compounds. X-Ray crystal structure of [Pd10(CO)12(O)6. J. Chem. Soc. Chem. Commun. 1981; 989–990. [Google Scholar] [CrossRef]
- Baranets, S.; Ovchinnikov, A.; Dmitrenko, O.; Bobev, S. Structural Uniqueness of the [Nb(As5)2]5- Cluster in the Zintl Phase Cs5NbAs10. J. Phys. Chem. A 2021, 125, 4323–4333. [Google Scholar] [CrossRef] [PubMed]
- Stroganova, E.A.; Troyanov, S.I.; Morozov, I.V.; Kuznetsov, A.N. Bismuth Polycations Revisited: Alternative Synthesis and Electronic Structure of Bi6Br7, and Bonding in Main-Group Polyatomic Ions from a Direct Space Perspective. Crystals 2020, 10, 940. [Google Scholar] [CrossRef]
- Bruker AXS Inc. SAINT 2014, Bruker AXS Inc.: Madison, Wisconsin, USA, 2014.
- Bruker AXS Inc. SADABS 2014, Bruker AXS Inc.: Madison, Wisconsin, USA, 2014.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Cryst. Struct. Commun. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Gelato, L.M.; Parthé, E. STRUCTURE TIDY—A Computer Program to Standardize Crystal Structure Data. J. Appl. Crystallogr. 1987, 20, 139–143. [Google Scholar] [CrossRef]
- Palmer, D.C. CrystalMaker; CrystalMaker Software Ltd.: Begbroke, Oxfordshire, UK, 2014. [Google Scholar]
- TURBOMOLE, V7.8. A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, TURBOMOLE GmbH 2024. Available online: http://www.turbomole.com (accessed on 15 November 2024).
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction, J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys. 2003, 119, 12129. [Google Scholar] [CrossRef]
- SGrimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets, J. Chem. Phys. 2015, 143, 054107. [Google Scholar]
- Hättig, C.; Schmitzand, J.G. Koßmann, Auxiliary basis sets for density-fitted correlated wavefunction calculations: Weighted core-valence and ECP basis sets for post-d elements. Phys. Chem. Chem. Phys. 2012, 14, 6549. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy. Phys. Chem. Chem. Phys/ 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Steffen, C.; Thomas, K.; Huniar, U.; Hellweg, A.; Rubner, O.; Schroer, A. TmoleX—A graphical user interface for TURBOMOLE. J. Comput. Chem. 2010, 31, 2967–2970. [Google Scholar] [CrossRef]
- Jepsen, O.; Andersen, O.K. The Stuttgart TB-LMTO-ASA Program; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1999. [Google Scholar]
- Barth, U.v.; Hedin, L. A Local Exchange-Correlation Potential for the Spin Polarized Case: I. J. Phys. C Solid State Phys. 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals 2018, 8, 225. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | [PdBi10][AlCl4]4 |
|---|---|
| fw/g mol−1 | 2871.32 |
| Space group | |
| a/Å | 11.0233(5) |
| b/Å | 26.1892(14) |
| c/Å | 26.2687(14) |
| a/° | 90.842(2) |
| β/° | 92.1940(10) |
| γ/° | 91.164(2) |
| V/Å3 | 7575.5(7) |
| Z | 8 |
| ρcalc./g cm–3 | 5.035 |
| Radiation | AgKα (λ = 0.56086) |
| μ(Ag-Kα)/mm–1 | 25.978 |
| 2θ range for data collection/° | 3.854 to 41.124 |
| Index ranges | −13 ≤ h ≤ 13, −32 ≤ k ≤ 32, −32 ≤ l ≤ 32 |
| Reflections collected/Independent reflections | 463,841/ 31,100 |
| R1 (I > 2σ(I)) [a] | 0.0360 |
| wR2 (I > 2σ(I)) [a] | 0.0694 |
| R1 (all data) [a] | 0.0428 |
| wR2 (all data) [a] | 0.0712 |
| Δρmax,min/e–·Å–3 | 2.848–(–2.020) |
| CCDC code | 2448552 |
| Bond Distance (Å)/Angle (°) | TPSSh | TPSSh with COSMO | M06-2X | B3-LYP | Experimental |
|---|---|---|---|---|---|
| d (Bi–Pd) | 3.0196–3.0212 | 2.9720–2.9828 | 3.0212–3.0228 | 3.0717–3.0736 | 2.9364(10)–3.0236(10) |
| d (Bi–Bi)Pentagon | 3.2036–3.2042 | 3.1434–3.1464 | 3.2117–3.2158 | 3.2645–3.2672 | 3.1168(7)–3.1964(7) |
| d (Bi–Bi)Triangle | 3.1001–3.1005 | 3.0785–3.0791 | 3.0798–3.0811 | 3.1359–3.1374 | 3.0656(9)–3.1552(9) |
| ∠(Bi–Bi–Bi)Pentagon | 107.95–108.03 | 107.98–108.02 | 107.96–108.01 | 107.94–108.05 | 105.08(2)–110.87(2) |
| ∠(Bi–Pd–Bi) | 61.75–61.78 | 63.84–63.88 | 61.30–64.22 | 61.36–61.40 | 62.05(2)–64.61(3) |
| ∠(Bi–Bi–Bi)Triangle | 58.88–62.23 | 59.27–61.46 | 58.55–62.87 | 58.61–62.76 | 57.812(16)–62.393(16) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samarakoon, S.M.G.K.; Baranets, S. [Palladium-decabismuth(4+)]-tetrakis(tetrachloridoaluminate) Cluster Compound, [Pd@Bi10][AlCl4]4: Synthesis, Crystal Structure, and Electronic Structure. Molbank 2025, 2025, M2020. https://doi.org/10.3390/M2020
Samarakoon SMGK, Baranets S. [Palladium-decabismuth(4+)]-tetrakis(tetrachloridoaluminate) Cluster Compound, [Pd@Bi10][AlCl4]4: Synthesis, Crystal Structure, and Electronic Structure. Molbank. 2025; 2025(2):M2020. https://doi.org/10.3390/M2020
Chicago/Turabian StyleSamarakoon, S. M. Gayomi K., and Sviatoslav Baranets. 2025. "[Palladium-decabismuth(4+)]-tetrakis(tetrachloridoaluminate) Cluster Compound, [Pd@Bi10][AlCl4]4: Synthesis, Crystal Structure, and Electronic Structure" Molbank 2025, no. 2: M2020. https://doi.org/10.3390/M2020
APA StyleSamarakoon, S. M. G. K., & Baranets, S. (2025). [Palladium-decabismuth(4+)]-tetrakis(tetrachloridoaluminate) Cluster Compound, [Pd@Bi10][AlCl4]4: Synthesis, Crystal Structure, and Electronic Structure. Molbank, 2025(2), M2020. https://doi.org/10.3390/M2020