
Diisopropyl (E)-(1-Hydroxy-3-phenylallyl)phosphonate


Abstract
1. Introduction
2. Results and Discussion
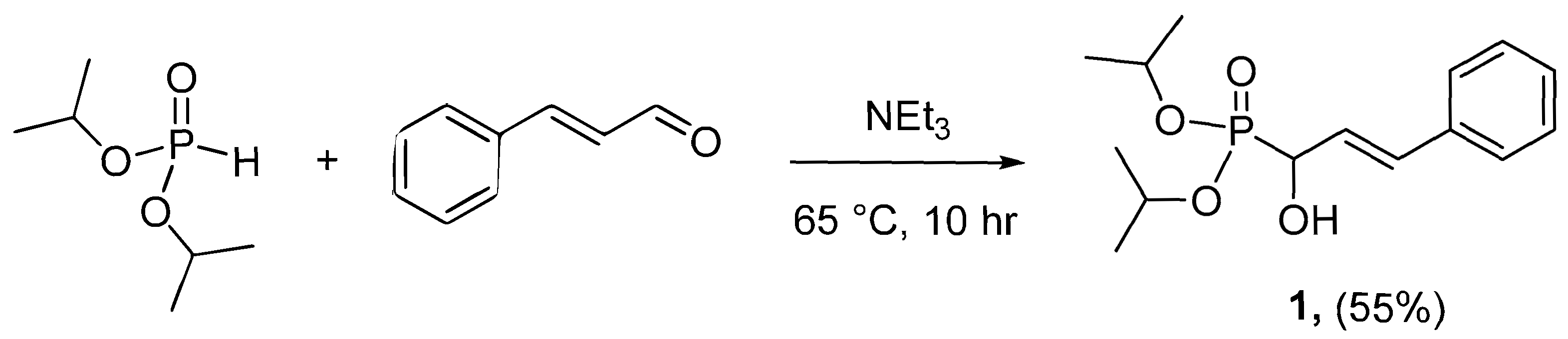
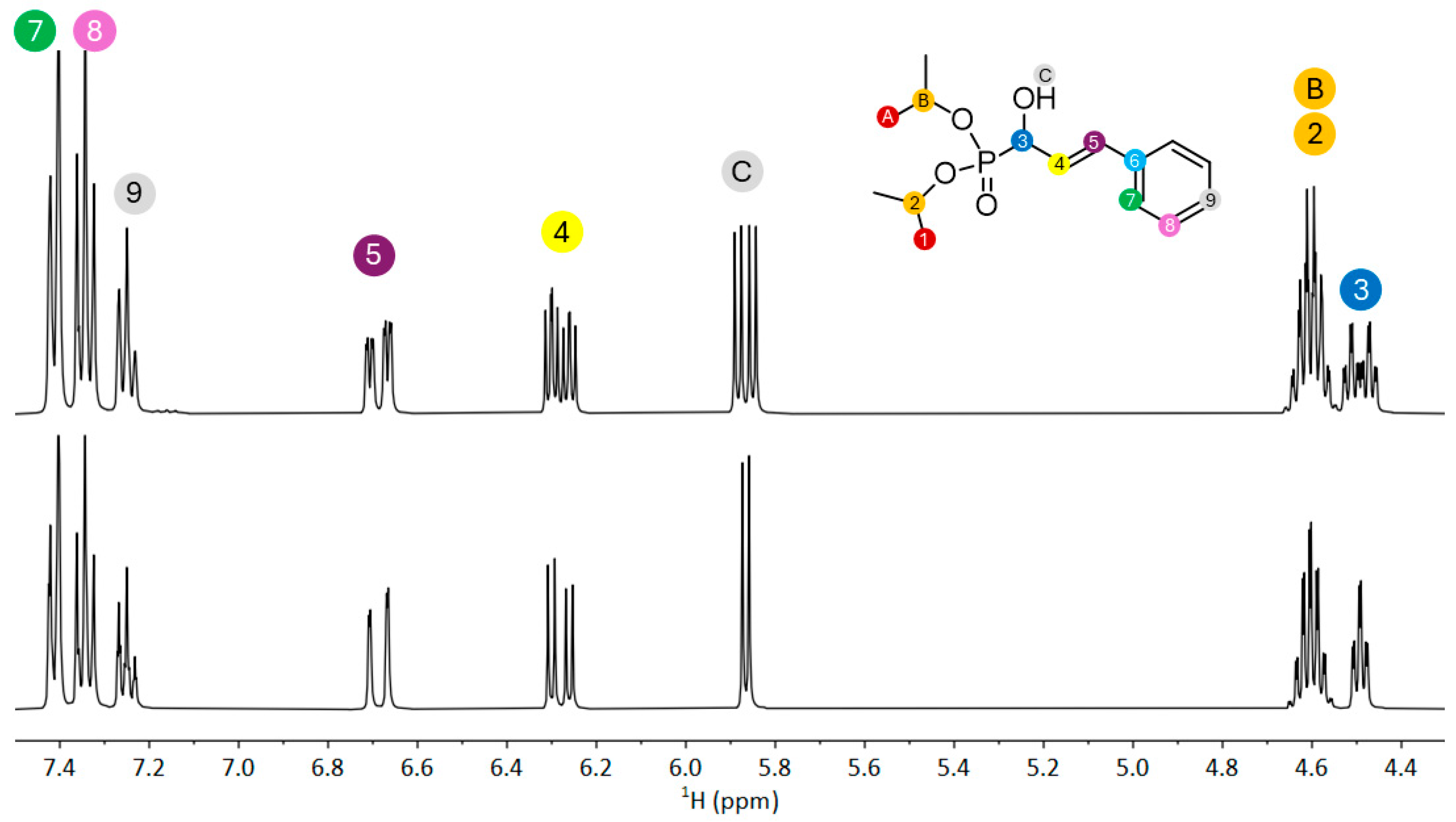
2.1. Synthesis and Spectroscopy
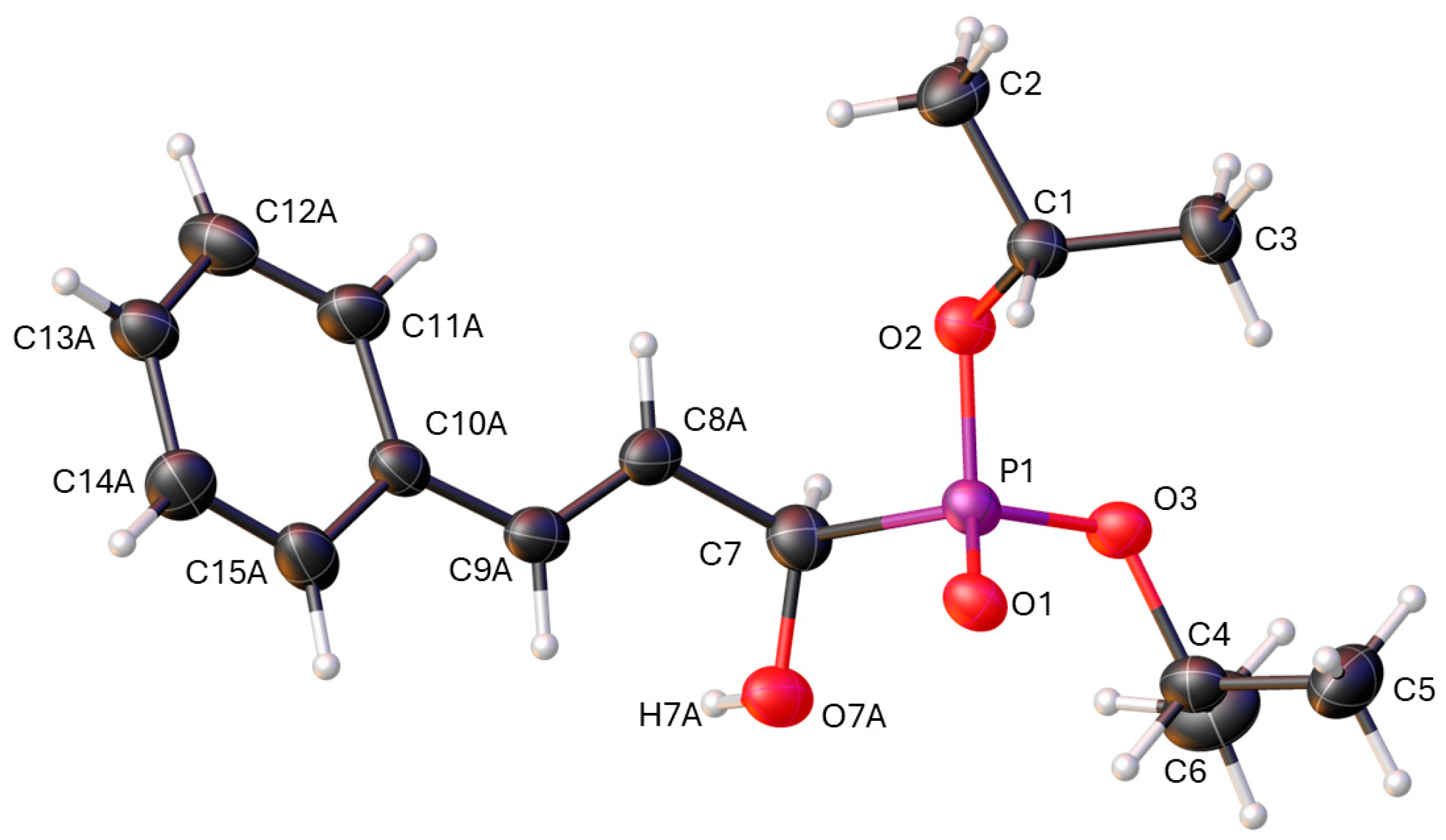
2.2. Structural Study
3. Materials and Methods
3.1. Synthesis and Characterisation of Diisopropyl (E)-(1-hydroxy-3-phenylallyl)phosphonate (1)
3.2. X-Ray Crystallography
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMR | Nuclear Magnetic Resonance |
| HRMS | High Resolution Mass Spectrometry |
| DMSO | dimethyl sulfoxide |
| ee | enantiomeric excess |
| vs | Very strong |
| m | Medium |
| w | Weak |
References
- Benech, J.M.; Coindet, M.; El Manouni, D.; Leroux, Y. Synthesis of New α-Ketophosphonates. Phosphours Sulfur Silicon 1997, 123, 377–383. [Google Scholar] [CrossRef]
- Rádai, Z. α-Hydroxyphosphonates as versatile starting materials. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 425–437. [Google Scholar] [CrossRef]
- Rádai, Z.; Keglevich, G. Synthesis and Reactions of α-Hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef]
- Pudovik, A.N. Addition of dialkyl phosphites to unsaturated compounds. A new method of synthesis of β-keto phosphonic and unsaturated α-hydroxyphosphonic esters. Doklady Akad. Nauk. SSSR 1950, 73, 449. [Google Scholar]
- Abell, J.P.; Yamamoto, H. Catalytic Enantioselective Pudovik Reaction of Aldehydes and Aldimines with Tethered Bis(8-quinolinato) (TBOx) Aluminum Complex. J. Am. Chem. Soc. 2008, 130, 10521–10523. [Google Scholar] [CrossRef]
- Fener, B.E.; Schuler, P.; Ueberschaar, N.; Bellstedt, P.; Görls, H.; Krieck, S.; Westerhausen, M. Scope and Limitations of the s-Block Metal-Mediated Pudovik Reaction. Chem. Eur. J. 2020, 26, 7235–7243. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Qian, D.-W.; Yang, S.-D. Lewis acid-catalysed Pudoivk reaction−phospha-Brook rearrangement sequence to access phosphoric esters. Beilsteon J. Org. Chem. 2022, 18, 1188–1194. [Google Scholar] [CrossRef]
- Yokomatsu, T.; Yamagishi, T.; Shibuya, S. Enantioselective synthesis of α-hydroxyphosphonates through asymmetric Pudovik reactions with chiral lanthanoid and titanium alkoxides. J. Chem. Soc. Perkin Trans. 1997, 1, 1527–1534. [Google Scholar] [CrossRef]
- Texier-Boullet, F.; Foucaud, A. A Convenient Synthesis of Dialkyl 1-Hydroxyalkanephosphonates using Potassium or Caesium Fluoride without Solvent. Synthesis 1982, 2, 165–166. [Google Scholar] [CrossRef]
- Smith, S.R.; Leckie, S.M.; Holmes, R.; Douglas, J.; Fallan, C.; Shapland, P.; Pryde, D.; Slawin, A.M.Z.; Smith, A.D. α-Ketophosphonates as Ester Surrogates: Isothiourea-Catalysed Asymmetric Diester and Lactone Synthesis. Org. Lett. 2014, 16, 2506–2509. [Google Scholar] [CrossRef]
- Öhler, E.; Kanzler, S. Synthesis of Phosphonic Acids Related to The Antibiotic Fosmidomycin From Allylic α- and γ- Hydroxyphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1996, 112, 71–74. [Google Scholar] [CrossRef]
- Leckie, S.M.; Fallan, C.; Taylor, J.E.; Brown, T.B.; Pryde, D.; Lébl, T.; Slawin, A.M.Z.; Smith, A.D. Enantiselective NHC-Catalysed Formal [4+2] Cycloaddition of Alkylarylketenes with β,γ-Unsaturated α-Ketophosphonates. Synlett 2013, 24, 1243–1249. [Google Scholar] [CrossRef]
- Meier, C.; Laux, W.H.G.; Bats, J.W. Asymmetric Synthesis of Chiral, Nonracemic Dialkyl α-Hydroxyarylmethyl- and α-, β- and γ-Hydroxyalkylphosphonates from Keto Phosphonates. Liebigs Ann. Recl. 1995, 11, 1936–1979. [Google Scholar] [CrossRef]
- Plouard, P.; Elmerich, U.; Hariri, M.; Loiseau, S.; Clarion, L.; Pirat, J.L.; Echeverria, P.-G.; Ayad, T.; Virieux, D. Ru(II)-Catalysed Enantioselective Transfer Hydrogenation of α-Ketophosphonates: Straightforward Access to Valuable Chiral α-Hydroxy Phosphonates. J. Org. Chem. 2023, 88, 16661–16665. [Google Scholar] [CrossRef]
- Goulioukina, N.S.; Bondaernko, G.N.; Gavrilov, K.N.; Beletskaya, I.P. Asymmetric Hydrogenation of α-Keto Phosphonates with Chiral Palladium Catalysts. Eur. J. Org. Chem. 2009, 2009, 510–515. [Google Scholar] [CrossRef]
- Zhao, Z.; Xue, W.; Gao, Y.; Tang, G.; Zhao, Y. Copper-Catalysed Synthesis of α-Hyroxy Phosphonates from H-Phosphonates and Alcohols or Ethers. Chem. Asian J. 2013, 8, 713–716. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z.; Kiss, N.Z. To date the greenest method for the preparation of α-hydroxyphosphonates from substituted bezaldehydes and dialkyl phosphites. Green. Process. Synth. 2017, 6, 197–201. [Google Scholar] [CrossRef]
- Kerim, M.D.; Katsina, T.; Cattoen, M.; Fincias, N.; Arseniyadis, S.; Kaïm, L.E. O-Allylated Pudovik and Passerini Adducts as Versatile Scaffolds for Product Diversification. J. Org. Chem. 2020, 85, 12514–12525. [Google Scholar] [CrossRef]
- McKay, A.P.; Cordes, D.B.; Smellie, I.A.; Chalmers, B.A. (E)-3-Mesityl-1-(2,3,4,5-tetramethylphenyl)prop-2-en-1-one. Molbank 2025, 2025, M1952. [Google Scholar] [CrossRef]
- Kühl, O. Phosphoros-31 NMR Spectroscopy: A Concise Introduction for the Synthetic Organic and Organometallic Chemist; Springer: Berlin/Heidelberg, Germany, 2008; pp. 7–22. [Google Scholar]
- Eslami, F.; Porayoubi, M.; Sabbaghi, F.; Dušek, M.; Baniyaghoob, S.; Skořepová, E. Database Survey of Single-and-Half Phosphorus–Oxygen Bonds in Salts with the C2PO2 Segment: Crystal Structure of [NH2C5H4NH][(C6H5)2P(O)(O)]·2H2O. Crystallogr. Rep. 2022, 67, 218–223. [Google Scholar] [CrossRef]
- Umezawa, Y.; Tsuboyama, S.; Honda, K.; Uzawa, J.; Nishio, M. CH/π Interaction in the Crystal Structure of Organic Compounds. A Database Study. Bull. Chem. Soc. Jpn. 1998, 71, 1207–1213. [Google Scholar] [CrossRef]
- CrysAlisPro, version 1.171.42.94a and 43.144a; Rigaku Oxford Diffraction, Rigaku Corporation: Tokyo, Japan, 2023.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1−O1 | 1.472(1) | C8A−C9A | 1.318(5) |
| P1−O2 | 1.566(1) | C7−O7A | 1.381(6) |
| P1−O3 | 1.574(1) | O1···H7A | 1.75(6) |
| P1−C7 | 1.822(2) | ||
| O1−P1−O2 | 115.50(7) | O2−P1−O3 | 103.34(6) |
| O1−P1−O3 | 113.98(7) | C7−P1−O2 | 101.02(7) |
| O1−P1−C7 | 114.51(8) | O1···H7A−O7A | 172(5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coker, A.I.; McKay, A.P.; Cordes, D.B.; Chalmers, B.A. Diisopropyl (E)-(1-Hydroxy-3-phenylallyl)phosphonate. Molbank 2025, 2025, M2013. https://doi.org/10.3390/M2013
Coker AI, McKay AP, Cordes DB, Chalmers BA. Diisopropyl (E)-(1-Hydroxy-3-phenylallyl)phosphonate. Molbank. 2025; 2025(2):M2013. https://doi.org/10.3390/M2013
Chicago/Turabian StyleCoker, Andy I., Aidan P. McKay, David B. Cordes, and Brian A. Chalmers. 2025. "Diisopropyl (E)-(1-Hydroxy-3-phenylallyl)phosphonate" Molbank 2025, no. 2: M2013. https://doi.org/10.3390/M2013
APA StyleCoker, A. I., McKay, A. P., Cordes, D. B., & Chalmers, B. A. (2025). Diisopropyl (E)-(1-Hydroxy-3-phenylallyl)phosphonate. Molbank, 2025(2), M2013. https://doi.org/10.3390/M2013