
Polymorphism of an Nα-Aroyl-N-Aryl-Phenylalanine Amide: An X-ray and Electron Diffraction Study

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
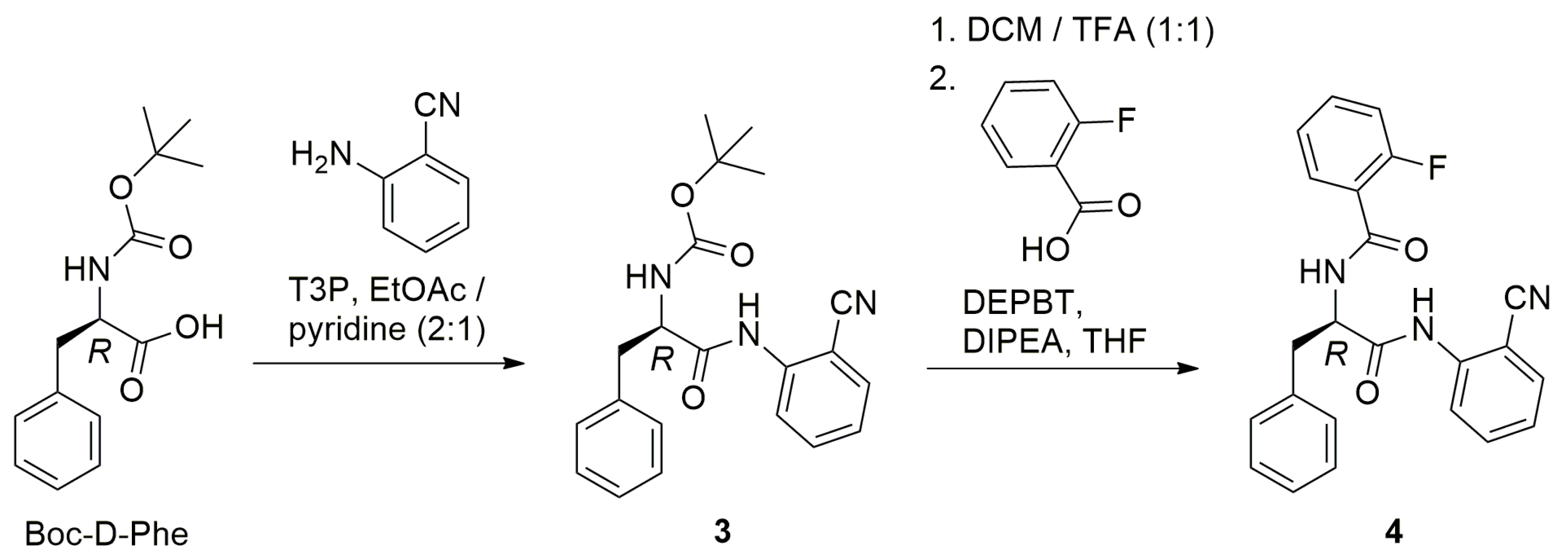
2.1. Synthesis
2.2. Structural Elucidation
2.3. Hirshfeld Surface Analysis and Density Functional Theory (DFT) Study
2.4. Antimycobacterial Evaluation
3. Materials and Methods
3.1. General
3.2. Synthesis
3.3. X-ray Crystallography
3.4. Electron Crystallography
3.5. Powder X-ray Diffraction
3.6. Computational Methods
3.7. Microbiological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; De Rosa, M.; Singla, N.; Singh, G.; Barnwal, R.P.; Pandey, A. Tuberculosis: An Overview of the Immunogenic Response, Disease Progression, and Medicinal Chemistry Efforts in the Last Decade toward the Development of Potential Drugs for Extensively Drug-Resistant Tuberculosis Strains. J. Med. Chem. 2021, 64, 4359–4395. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Herrmann, J.L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 2020, 18, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Kumar, S.; Singh, V. Unlocking Opportunities for Mycobacterium leprae and Mycobacterium ulcerans. ACS Infect. Dis. 2024, 10, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Farhat, M.; Cox, H.; Ghanem, M.; Denkinger, C.M.; Rodrigues, C.; Abd El Aziz, M.S.; Enkh-Amgalan, H.; Vambe, D.; Ugarte-Gil, C.; Furin, J.; et al. Drug-resistant tuberculosis: A persistent global health concern. Nat. Rev. Microbiol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Mirzayev, F.; Cirillo, D.M.; Udwadia, Z.; Dooley, K.E.; Chang, K.C.; Omar, S.V.; Reuter, A.; Perumal, T.; Horsburgh, C.R., Jr.; et al. Multidrug-resistant tuberculosis. Nat. Rev. Dis. Primers 2024, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Prevots, D.R.; Marshall, J.E.; Wagner, D.; Morimoto, K. Global Epidemiology of Nontuberculous Mycobacterial Pulmonary Disease: A Review. Clin. Chest Med. 2023, 44, 675–721. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V.; Dick, T. Therapeutic developments for tuberculosis and nontuberculous mycobacterial lung disease. Nat. Rev. Drug Discov. 2024, 23, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Seidel, R.W.; Goddard, R.; Lang, M.; Richter, A. Nα-aroyl-N-aryl-phenylalanine amides: A promising class of antimycobacterial agents targeting the RNA polymerase. Chem. Biodivers. 2024, 21, e202400267. [Google Scholar] [CrossRef]
- Lin, W.; Mandal, S.; Degen, D.; Liu, Y.; Ebright, Y.W.; Li, S.; Feng, Y.; Zhang, Y.; Mandal, S.; Jiang, Y.; et al. Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol. Cell 2017, 66, 169–179. [Google Scholar] [CrossRef]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; Gonzalez, R.; et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Low, J.L.; Wu, M.-L.; Aziz, D.B.; Laleu, B.; Dick, T. Screening of TB Actives for Activity against Nontuberculous Mycobacteria Delivers High Hit Rates. Front. Microbiol. 2017, 8, 1539. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, G.; Moon, C.; Kim, H.J.; Kim, T.H.; Jang, J. Pathogen Box screening for hit identification against Mycobacterium abscessus. PLoS ONE 2018, 13, e0195595. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Strauch, A.; Chao, J.; Ko, M.; Av-Gay, Y. Screening of Preselected Libraries Targeting Mycobacterium abscessus for Drug Discovery. Antimicrob. Agents Chemother. 2018, 62, e00828-18. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Shapira, T.; Av-Gay, Y. THP-1 and Dictyostelium Infection Models for Screening and Characterization of Anti-Mycobacterium abscessus Hit Compounds. Antimicrob. Agents Chemother. 2019, 64, e01601-19. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Ganapathy, U.S.; Mann, L.; Abdelaziz, R.; Seidel, R.W.; Goddard, R.; Sequenzia, I.; Hoenke, S.; Schulze, P.; Aragaw, W.W.; et al. Synthesis and Characterization of Phenylalanine Amides Active against Mycobacterium abscessus and Other Mycobacteria. J. Med. Chem. 2023, 66, 5079–5098. [Google Scholar] [CrossRef]
- Lang, M.; Ganapathy, U.S.; Mann, L.; Seidel, R.W.; Goddard, R.; Erdmann, F.; Dick, T.; Richter, A. Synthesis and in vitro Metabolic Stability of Sterically Shielded Antimycobacterial Phenylalanine Amides. ChemMedChem 2024, 19, e202300593. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, H.; Kleiner, H.J. New Peptide Synthesis. Angew. Chem.-Int. Ed. Engl. 1980, 19, 133–134. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Xiang, Y.; Baldwin, A.; Ringling, J. General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine. Org. Lett. 2011, 13, 5048–5051. [Google Scholar] [CrossRef]
- Waghmare, A.A.; Hindupur, R.M.; Pati, H.N. Propylphosphonic anhydride (T3P®): An expedient reagent for organic synthesis. Rev. J. Chem. 2014, 4, 53–131. [Google Scholar] [CrossRef]
- Waring, A.; Joullie, M.M.; Lassen, K.M. Evolution of amide bond formation. Arkivoc 2010, 2010, 189–250. [Google Scholar] [CrossRef]
- Ye, Y.H.; Li, H.; Jiang, X. DEPBT as an efficient coupling reagent for amide bond formation with remarkable resistance to racemization. Biopolymers 2005, 80, 172–178. [Google Scholar] [CrossRef]
- Li, H.; Jiang, X.; Ye, Y.-h.; Fan, C.; Romoff, T.; Goodman, M. 3-(Diethoxyphosphoryloxy)-1,2,3- benzotriazin-4(3H)-one (DEPBT): A New Coupling Reagent with Remarkable Resistance to Racemization. Org. Lett. 1999, 1, 91–94. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Kitajgorodskij, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic-compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Yee, M.; Klinzing, D.; Wei, J.R.; Gengenbacher, M.; Rubin, E.J.; Dick, T. Draft Genome Sequence of Mycobacterium abscessus Bamboo. Genome Announc. 2017, 5, e00388-17. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Midgley, L.; Bourhis, L.J.; Dolomanov, O.V.; Grabowsky, S.; Kleemiss, F.; Puschmann, H.; Peyerimhoff, N. Vanishing of the atomic form factor derivatives in non-spherical structural refinement—A key approximation scrutinized in the case of Hirshfeld atom refinement. Acta Crystallogr. A 2021, 77, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Bernstein, H.J.; Hammersley, A.P. Specification of the Crystallographic Binary File (CBF/imgCIF). In International Tables for Crystallography Volume G: Definition and Exchange of Crystallographic Data; Hall, S.R., McMahon, B., Eds.; Springer: Dordrecht, The Netherlands, 2006; Chapter 2.3; pp. 37–43. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Klar, P.B.; Krysiak, Y.; Xu, H.; Steciuk, G.; Cho, J.; Zou, X.; Palatinus, L. Accurate structure models and absolute configuration determination using dynamical effects in continuous-rotation 3D electron diffraction data. Nat. Chem. 2023, 15, 848–855. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d(D–H) | d(H···A) | d(D···A) | <(DHA) | |
|---|---|---|---|---|
| N1–H1···O1a | 0.99 (3) | 2.05 (3) | 2.935 (3) | 146 (2) |
| N2–H2A···O2b | 0.98 (3) | 1.80 (3) | 2.777 (3) | 169 (2) |
| C16–H16···N3b | 1.10 (3) | 2.42 (3) | 3.491 (3) | 165 (2) |
| d(D–H) | d(H···A) | d(D···A) | <(DHA) | |
|---|---|---|---|---|
| N1–H1···O2 | 1.03 (3) | 2.14 (5) | 2.98 (2) | 137 (4) |
| N2–H2A···O2a | 1.02 (3) | 2.10 (3) | 3.06 (2) | 156 (4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lang, M.; Goddard, R.; Patzer, M.; Ganapathy, U.S.; Dick, T.; Richter, A.; Seidel, R.W. Polymorphism of an Nα-Aroyl-N-Aryl-Phenylalanine Amide: An X-ray and Electron Diffraction Study. Molbank 2024, 2024, M1851. https://doi.org/10.3390/M1851
Lang M, Goddard R, Patzer M, Ganapathy US, Dick T, Richter A, Seidel RW. Polymorphism of an Nα-Aroyl-N-Aryl-Phenylalanine Amide: An X-ray and Electron Diffraction Study. Molbank. 2024; 2024(3):M1851. https://doi.org/10.3390/M1851
Chicago/Turabian StyleLang, Markus, Richard Goddard, Michael Patzer, Uday S. Ganapathy, Thomas Dick, Adrian Richter, and Rüdiger W. Seidel. 2024. "Polymorphism of an Nα-Aroyl-N-Aryl-Phenylalanine Amide: An X-ray and Electron Diffraction Study" Molbank 2024, no. 3: M1851. https://doi.org/10.3390/M1851
APA StyleLang, M., Goddard, R., Patzer, M., Ganapathy, U. S., Dick, T., Richter, A., & Seidel, R. W. (2024). Polymorphism of an Nα-Aroyl-N-Aryl-Phenylalanine Amide: An X-ray and Electron Diffraction Study. Molbank, 2024(3), M1851. https://doi.org/10.3390/M1851