
N-(diisopropylphosphanyl)benzamide



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
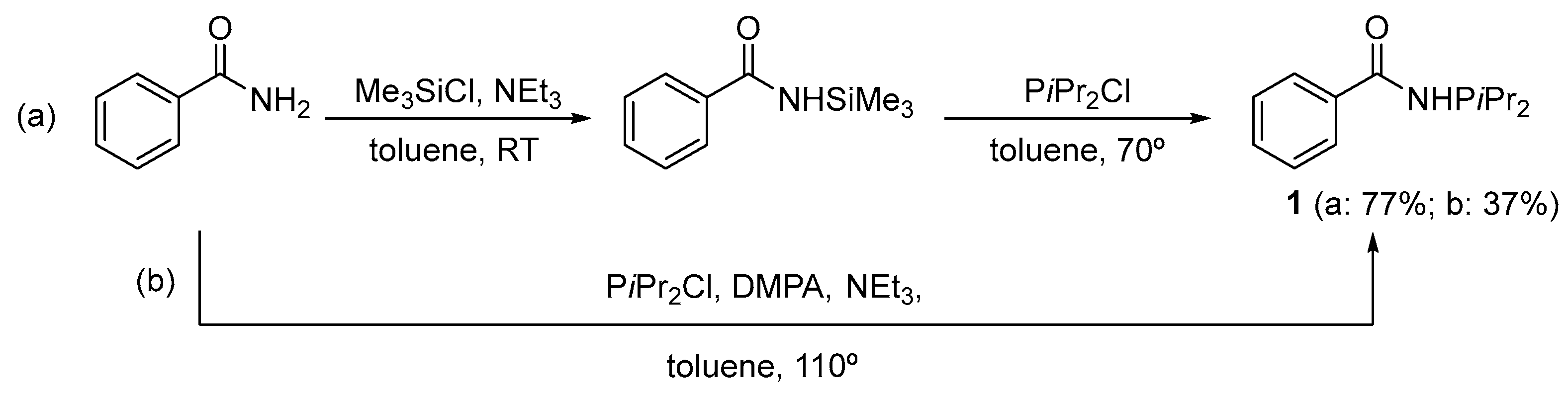
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pearson, R.G. Hard and soft acid and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Jeffrey, J.C.; Rauchfuss, T.B. Metal complexes of hemilabile ligands. Reactivity and structure of dichlorobis(o-(diphenylphosphino)anisole)ruthenium(II). Inorg. Chem. 1979, 18, 2658–2666. [Google Scholar] [CrossRef]
- Bader, A.; Lindner, E. Coordination chemistry and catalysis with hemilabile oxygen-phosphorus ligands. Coord. Chem. Rev. 1991, 108, 27–110. [Google Scholar] [CrossRef]
- Braunstein, P. Functional ligands and complexes for new structures, homogeneous catalysts and nanomaterials. J. Organomet. Chem. 2004, 689, 3953–3967. [Google Scholar] [CrossRef]
- Braunstein, P.; Naud, F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem. Int. Ed. 2001, 40, 680–699. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X.; Adams, R.D. Coordination properties of novel hemilabile acetamide-derived P,O phosphine ligands. Crystal structures of Ph2PNHC(O)Me and [PdMe{PPh2NHC(O)Me}{PPh2NHC(O)Me}][O3SCF3]. J. Chem. Soc. Dalton Trans. 2000, 13, 2205–2214. [Google Scholar] [CrossRef]
- Milton, H.L.; Wheatley, M.V.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and coordination of 2-diphenylphosphinopicolinamide. Polyhedron 2004, 23, 3211–3220. [Google Scholar] [CrossRef]
- Ly, T.Q.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and coordination chemistry of RC(E)NHPPh2 (E = O or S; R = H2N, Ph, or Py). X-ray structure of [Pt{H2NC(S)NHPPh2}2] 2Cl 0.5MeOH. Polyhedron 1999, 18, 1761–1766. [Google Scholar] [CrossRef]
- Morise, X.; Green, M.L.H.; Braunstein, P.; Rees, L.H.; Vei, I.C. Monocyclopentadienyl complexes of niobium, tantalum and tungsten containing heterodifunctional P,O ligands. New J. Chem. 2003, 27, 32–38. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X. Synthesis and crystal structure of [PdMe{PPh2NHC(O)Me}{O=C(NH2)Me}][BF4], a palladium complex containing the acetamide ligand. C. R. Chimie 2002, 5, 131–135. [Google Scholar] [CrossRef]
- Oberbeckmann-Winter, N.; Braunstein, P.; Welter, R. Palladium Phosphino-Iminolate Complexes as Metalloligands for Coinage Metals: A Versatile, Ambivalent Behavior. Organometallics 2005, 24, 3149–3157. [Google Scholar] [CrossRef]
- Jones, N.G.; Green, M.L.H.; Vei, I.; Cowley, A.; Morise, X.; Braunstein, P. Synthesis of molybdenum arene complexes containing amide-derived heterodifunctional P,O ligands. J. Chem. Soc. Dalton Trans. 2002, 1487–1493. [Google Scholar] [CrossRef]
- Agostinho, M.; Rosa, V.; Avilés, T.; Welter, R.; Braunstein, P. Synthesis and characterization of Co and Ni complexes stabilized by keto- and acetamide-derived P,O-type phosphine ligands. Dalton Trans. 2009, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Navratil, M.; Cisarova, I.; Stepnicka, P. Intermolecular interactions in the crystal structures of chlorogold(I) complexes with N-phosphinoamide ligands. Inorg. Chim. Acta 2021, 516, 120138. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Ly, T.Q.; Slawin, A.M.Z.; Woollins, J.D. P- versus P,O- coordination in complexes of N-(diphenyl phosphino)arylamide ligands ArC(O)NHPPh2 (Ar = 3-pyridyl, phenyl). X-ray crystal structures of [RhCl2(η5-C5Me5)L2] and [NiCl(EtOH)L22]·Cl·[NiCl2L22] (L2= C6H5CONHPPh2). Polyhedron 2001, 20, 1803–1808. [Google Scholar] [CrossRef]
- Braunstein, P.; Frison, C.; Morise, X. Stepwise ethene and/or methyl acrylate/CO insertions into the Pd-C bond of cationic palladium(II) complexes stabilized by a (P,O) chelate. Angew. Chem. Int. Ed. 2000, 39, 2867–2870. [Google Scholar] [CrossRef]
- Li, T.-Y.; Liang, X.; Wu, C.; Xue, L.-S.; Xu, Q.-L.; Zhang, S.; Liu, X.; Zheng, Y.-X.; Wang, X.-Q. Syntheses, crystal structure, photophysical property and theoretical study of a new series of iridium complexes with N-(diphenylphosphoryl)benzamide derivatives as the ancillary ligands. J. Organomet. Chem. 2014, 755, 110–119. [Google Scholar] [CrossRef]
- Blondiaux, E.; Cantat, T. Efficient metal-free hydrosilylation of tertiary, secondary and primary amides to amines. Chem. Commun. 2014, 50, 9349–9352. [Google Scholar] [CrossRef]
- Bruker. SAINT. In APEX2; Bruker AXS Inc.: Madison, WI, USA, 2007. [Google Scholar]
- Sheldrick, G.M. SADABS, Programs for Scaling and Absorption Correction of Area Detector Data; SADABS, Programs Scaling Absorpt. Correct. Area Detect. Data; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Burla, M.C.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2002: The Program. J. Appl. Cryst. 2003, 36, 1103. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcaide, M.M.; Garrido, D.G.; Álvarez, E.; Peloso, R. N-(diisopropylphosphanyl)benzamide. Molbank 2023, 2023, M1667. https://doi.org/10.3390/M1667
Alcaide MM, Garrido DG, Álvarez E, Peloso R. N-(diisopropylphosphanyl)benzamide. Molbank. 2023; 2023(2):M1667. https://doi.org/10.3390/M1667
Chicago/Turabian StyleAlcaide, María M., Daniel García Garrido, Eleuterio Álvarez, and Riccardo Peloso. 2023. "N-(diisopropylphosphanyl)benzamide" Molbank 2023, no. 2: M1667. https://doi.org/10.3390/M1667
APA StyleAlcaide, M. M., Garrido, D. G., Álvarez, E., & Peloso, R. (2023). N-(diisopropylphosphanyl)benzamide. Molbank, 2023(2), M1667. https://doi.org/10.3390/M1667