1. Introduction
Since
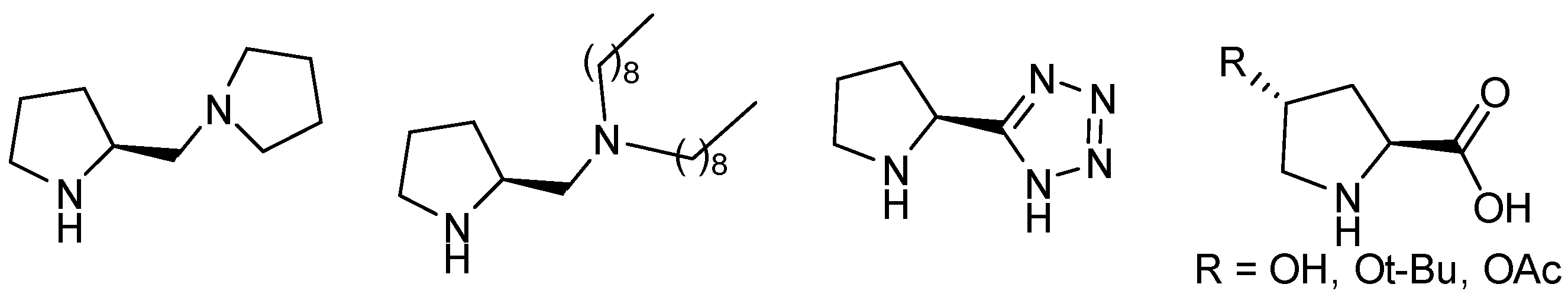
l-Proline (
l-Pro) was employed as chiral organocatalysts in the asymmetric aldol reaction in 1999 by Barbas and List [
1], many pyrrolidine derivatives based on this amino acid have been synthesized and used in asymmetric organocatalysis. Most of these derivatives are based on different modifications of the carboxylic acid of
l-Pro. Some of these modifications consist of transforming this group into a pyrrolidine ring [
2], a highly lipophilic tertiary amine [
3] or a tetrazole [
4] (
Figure 1), leading to active organocatalysts, even when functionalizing the pyrrolidine ring, as in
trans-4-hydroxi-
l-proline and derivatives (
Figure 1) [
5].
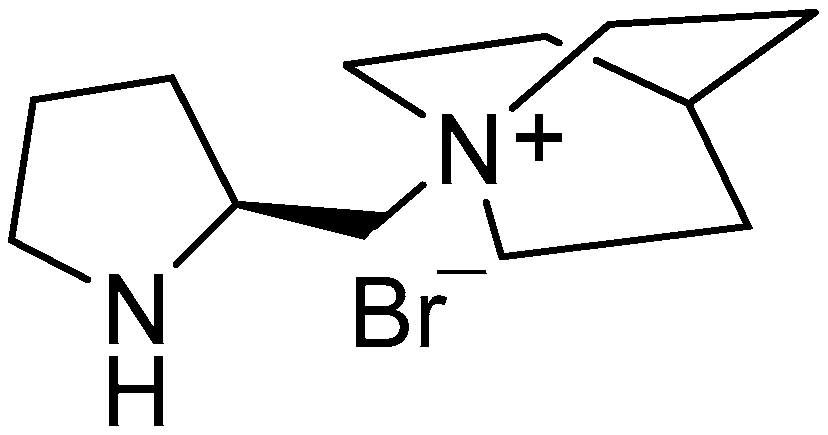
Different alkylammonium salts based on
l-Pro have also been shown as efficient organocatalysts in asymmetric Michael additions of ketones to
trans-β-nitrostyrenes [
6]. Some of these salts have been recycled three times after the Michael addition without loss of enantioselectivity [
7] (
Figure 2).
In addition, many chiral quaternary ammonium salts have been employed as catalysts in other areas of organocatalysis, such as asymmetric phase-transfer catalysis (PTC) [
8]. These types of catalysts facilitate the transfer of compounds from one reaction phase to another, quaternary ammonium salts derived from
Cinchona alkaloids being the most employed [
9] as Corey’s
O-(9)-allyl-
N-(9-anthracenylmethyl)-cinchonidinium bromide (
Figure 3), which has been successfully used in the asymmetric alkylation of enolate glycinates [
10].
In this communication, we report the synthesis of the new
l-Pro-derived chiral ammonium salt (
S)-(1-pyrrolidin-2-ylmethyl)quinuclidine-1-ium bromide (
Figure 4), which has been spectroscopically and thermally characterized.
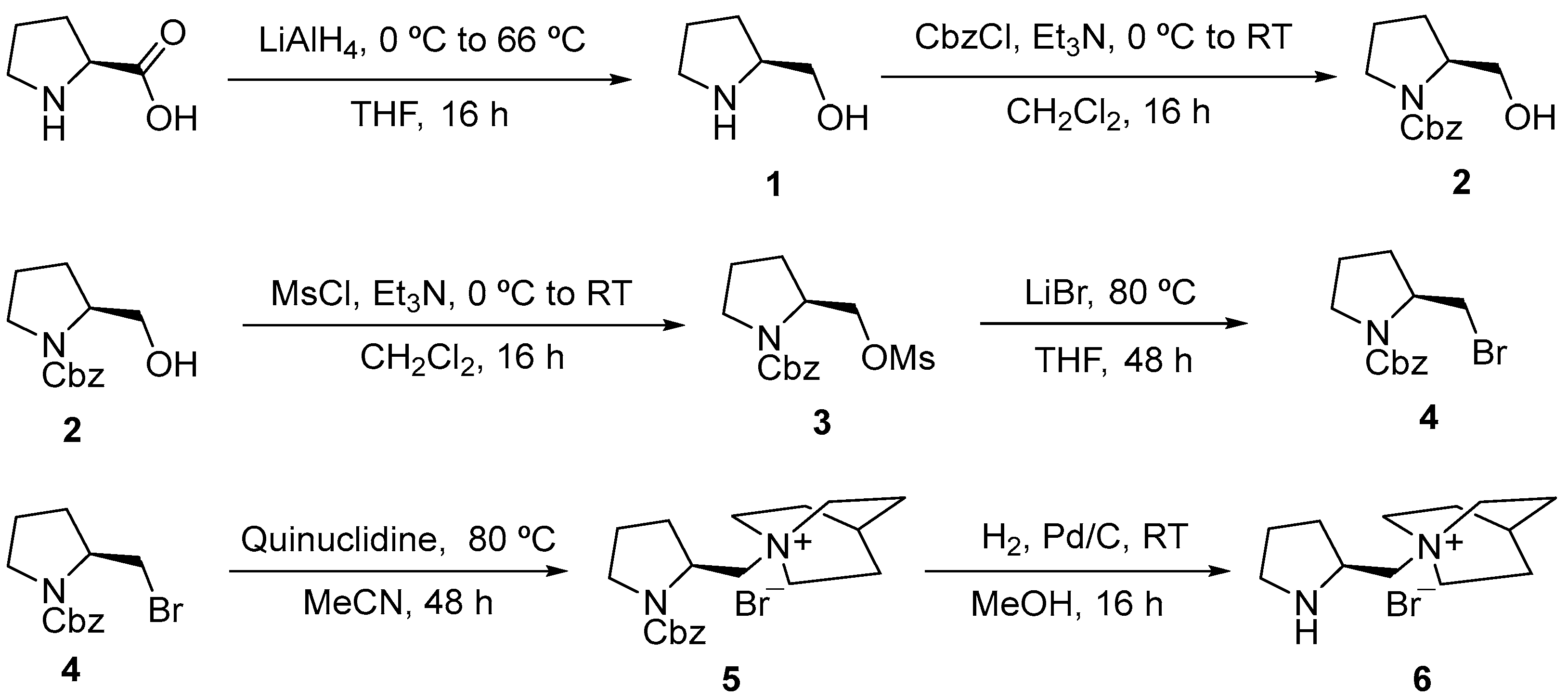
2. Results
The synthesis of (
S)-(1-pyrrolidin-2-ylmethyl)quinuclidine bromide (
6) was carried out using a six-step reaction pathway, as shown in
Scheme 1.
l-Proline was initially reduced to
l-Prolinol (
1) with LiAlH
4. Then,
1 was
N-Cbz protected with benzyl chloroformate at 0 °C to RT, giving product
2 as an orange oil, its structure being verified by the aromatic and benzylic signals in the
1H-NMR (300 MHz, CDCl
3) spectrum (see
Supplementary Materials). Then, the hydroxy group of
2 was mesylated with methanesulfonyl chloride to give compound
3 as a viscous brown oil, its structure being confirmed (see
Supplementary Materials) by
1H-NMR (300 MHz, CDCl
3), which shows the corresponding spectrum a signal at 2.84–2.94 (m, 3H, SCH
3). Subsequently, compound
4 was obtained as a dense brown oil after nucleophilic substitution over
3 with LiBr and further purification by column chromatography. The structure of this bromide
4 was confirmed by
1H-NMR (300 MHz, CDCl
3), which showed a signal, among others, at 4.13 (br s, 1H, C
HNCbz) (see
Supplementary Materials). So far, all the reaction steps were carried out using the crude intermediate compounds, since high yields and purities were always observed (
Table 1), to reduce solvent consumption. The first attempt to produce
5 was based on a nucleophilic substitution with quinuclidine at RT (16 h), but the yield was pretty low. Therefore, the next attempt was to extend the reaction time to 48 h at 80 °C, which led to, after three washes with EtOAc and two with
n-hexane to remove impurities of previous steps, compound
5 as a slightly orange solid. The final step towards
6 was a hydrogenolysis of
5 to remove the Cbz. The procedure consisted of carrying out a 16 h reduction of
5 under H
2 atmosphere (1 atm) and Pd/C (10%) as catalyst. This reaction was repeated four times by adding fresh catalyst after filtration of the crude reaction to obtain complete conversion (
1H-NMR spectrum of the crude product). After three washes with EtOAc and two with
n-hexane, the final product
6 was obtained and characterized by
1H-NMR,
13C-NMR, LRMS-HPLC, TGA-DSC and IR-ATR (see
Supplementary Materials).
As described, purifications were not performed by column chromatography due to the complexity of separating compounds 4–6 from the impurities using this technique and to reduce solvent consumption. Instead, the crude reaction mixtures were washed with organic solvents to remove the impurities. Among all the tested solvents, diethyl ether, CH2Cl2 and toluene were unsuccessful, with EtOAc and n-hexane being the most efficient ones at removing the impurities and the unreactive reagents, such as quinuclidine.
3. Experimental
Unless otherwise noted, all commercial reagents and solvents were used without further purification. Reactions under an argon atmosphere were carried out in oven-dried glassware sealed with rubber septum using anhydrous solvents. Melting points were determined using a Reichert Thermovar microscope (Vienna, Austria) and are uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded for 1H-NMR at 300 or 400 MHz and for 13C-NMR at 75 or 101 MHz on a Bruker AC-300 or AC-400 instrument from the Technical Services (SSTTI) of the University of Alicante (UA), using CDCl3 as solvent and TMS (0.0003%) as internal standard, unless otherwise indicated. Chemical shifts (δ) were expressed in units of parts per million relative to TMS and coupling constant (J) in Hertz (Hz). The multiplicity of signals was indicated as follows: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), m (multiplet). Low-resolution mass spectrometry coupled to HPLC (LRMS-HPLC) analyses were performed on an Agilent Model 1100 Series High-Performance Liquid Chromatograph coupled to a variable wavelength visible-UV detector and a mass spectrometer with an ion trap spectroscopy analyzer at the SSTTI of the UA. Infrared spectra (IR) were recorded on a JASCO FT/IR 4100 spectrometer equipped with an ATR component; wavenumbers were expressed in cm−1. Thin layer chromatography (TLC) was performed on precast chromatoplates on silica gel 60 (Merck silica gel 60 F254). Development of TLC plates was carried out either using a UV lamp irradiating at a wavelength of 254 nm, using I2 vapors, or by oxidation with 30% phosphomolybdic acid in ethanol. Column chromatography (CC) was performed in glass columns using Merck silica gel 60, with a particle size of 0.060–0.200 nm as the stationary phase. Differential scanning calorimetry (DSC) was carried out at the SSTTI of the UA in a TA Instruments MDSC temperature modulation heat flow DSC unit, model Q250, performing two heating cycles and one cooling cycle. Thermogravimetric tests (TGA) were carried out at the SSTTI of the UA using a METTLER-TOLEDO TG-DSC2 instrument equipped with an automatic changer and a humidity control module. Experimental synthesis and spectroscopic data are provided starting from product 4.
Benzyl (S)-2-(bromomethyl)pyrrolidine-1-carboxylate (4). To a stirring solution under Argon atmosphere of compound 2 (12.6 g, 40.3 mol) in dry THF (50 mL), LiBr (6.6 g, 86.8 mmol, 2.5 equiv.) was portion-wise added. The reaction mixture was stirred for 16 h at reflux. Then, THF was evaporated under vacuum, 150 mL of H2O was added and three extractions with EtOAc (3 × 150 mL) were performed. The collected and combined organic phases were dried with anhydrous Na2SO4, and after filtration, the solvent was evaporated under vacuum, leaving a residue that was purified by flash chromatography using an EtOAc/Hx: 1/4 mixture as eluent to led to compound 3 (8.1 g, 67%, 27 mmol) as a heavy brown oil. 1H-NMR (300 MHz, CDCl3), δH (mixture of rotamers): 1.83–2.09 (m, 4H, -CH2CH2-), 3.28–3.71 (m, 4H, CH2NCbz y CH2Br), 4.13 (br s, 1H, CHNCbz), 5.13 (d, J = 12 Hz, 1H, CHHPh) and 5.21 (d, J = 12 Hz, 1H, CHHPh), 7.32–7.37 (m, 5H, ArH) ppm. 13C-NMR (75 MHz, CDCl3), δC (mixture of rotamers): 22.8, 23.6, 29.4, 30.1, 34.7, 47.2, 47.5, 57.8, 58.2, 66.8, 67.0, 127.9, 128.0, 128.1, 128.5, 128.6, 136.6, 136.7, 154.9 ppm.
(S)-1-((1-((Benzyloxy)carbonyl)pyrrolidin-2-yl)-methyl)quinuclidinium bromide (5). Under Argon atmosphere, product 3 (8.1 g, 27 mmol) was dissolved in MeCN (100 mL), and quinuclidine (3 g, 27 mmol, 1 equiv.) was portion-wise added. The reaction mixture was heated at 80 °C for 48 h. The solvent was then evaporated under vacuum to yield a crunchy orange solid that was washed three times with EtOAc (3 × 50 mL) and two times with n-hexane (2 × 50 mL) to remove the impurities from the previous synthetic steps. Product 4 (7.85 g, 71%, 19.2 mmol) was obtained as a slightly orange solid after drying in vacuo. 1H-NMR (300 MHz, MeOH-d4), δH (mixture of rotamers): 1.75–1.83 (m, 2H), 1.88–2.00 (m, 9H), 2.06–2.23 (m, 2H), 3.23–3.27 (m, 2H), 3.42–3.62 (m, 6H), 4.33–4.45 (m, 1H, CHNCbz), 5.14 (br s, 2H, CH2Ph), 7.28–7.48 (m, 5H, ArH) ppm. 13C-NMR is not given due to the spectra’s complexity because of the rotamers’ mixture.
(S)-(1-Pyrrolidin-2-ylmethyl)quinuclidine-1-ium bromide (6). To a 250 mL round bottom flask containing compound 5 (7.85 g, 19.2 mmol) and Pd/C (1 g) under Argon atmosphere, MeOH (50 mL) was added. After compound 5 was completely dissolved, the reaction atmosphere was changed to hydrogen (1 atm) by 3 vacuum/hydrogen cycles, and the resulting mixture was stirred for 16 h at room temperature. After this time, the resulting mixture was filtered through celite to remove Pd/C, and MeOH was evaporated in vacuo to yield the crude product as an orange solid. The crude product was not completely deprotected, so the hydrogenolysis process was repeated four more times. After checking by 1H-NMR that the reaction was completed, the crude product was washed with EtOAc (3 × 50 mL) and n-hexane (2 × 50 mL) to remove the impurities. Then, the product was dried in an oven (60 °C) for two days to yield pure salt 6 (3.73 g, 70%, 13.6 mmol) as a slightly yellowish-white solid. Melting point 190 °C (MeOH). 1H-NMR (300 MHz, MeOH-d4), δH: 1.37–1.5 (m, 1H, -CH2CHCH2-), 1.7–2.19 (m, 11H), 2.96–3.01 (m, 2H, -CH2NH-), 3.19–3.27 (m, 1H), 3.33–3.37 (m, 1H), 3.48–3.73 (m, 7H) ppm. 13C-NMR (75 MHz, MeOH-d4), δC: 21.0, 24.9, 25.9, 32.9, 47.7, 54.1, 56.6, 69.2 ppm. m/z 195.2 (positive ion).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}