
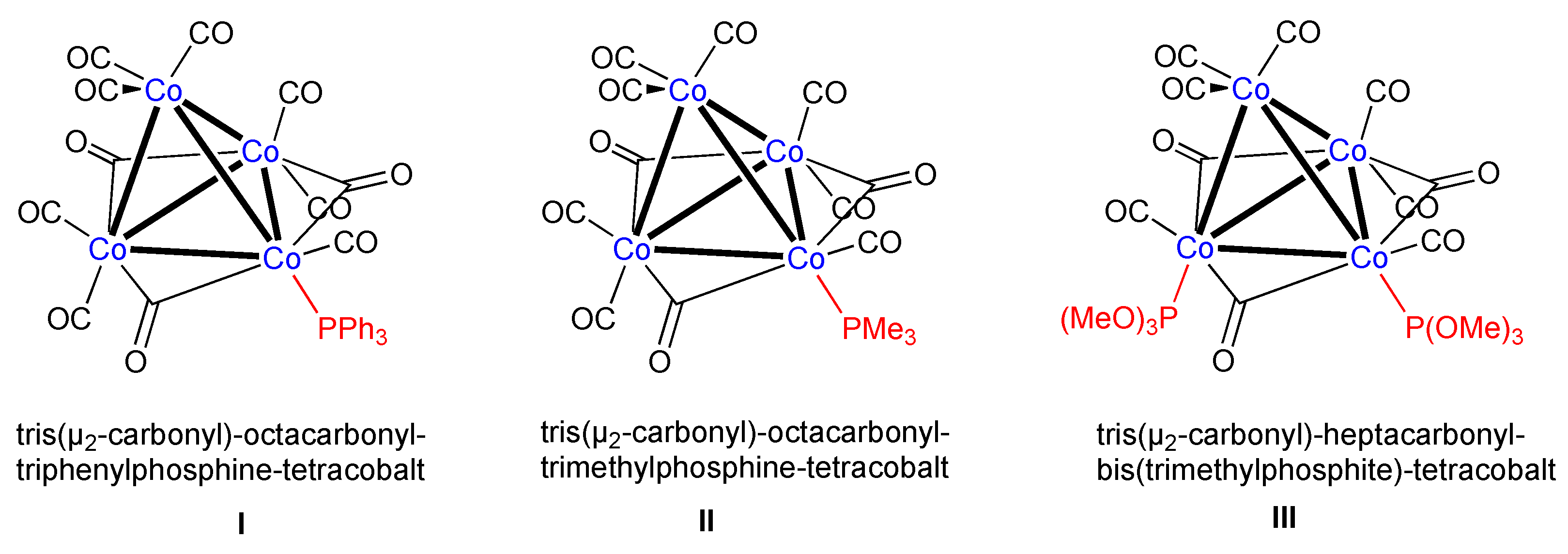
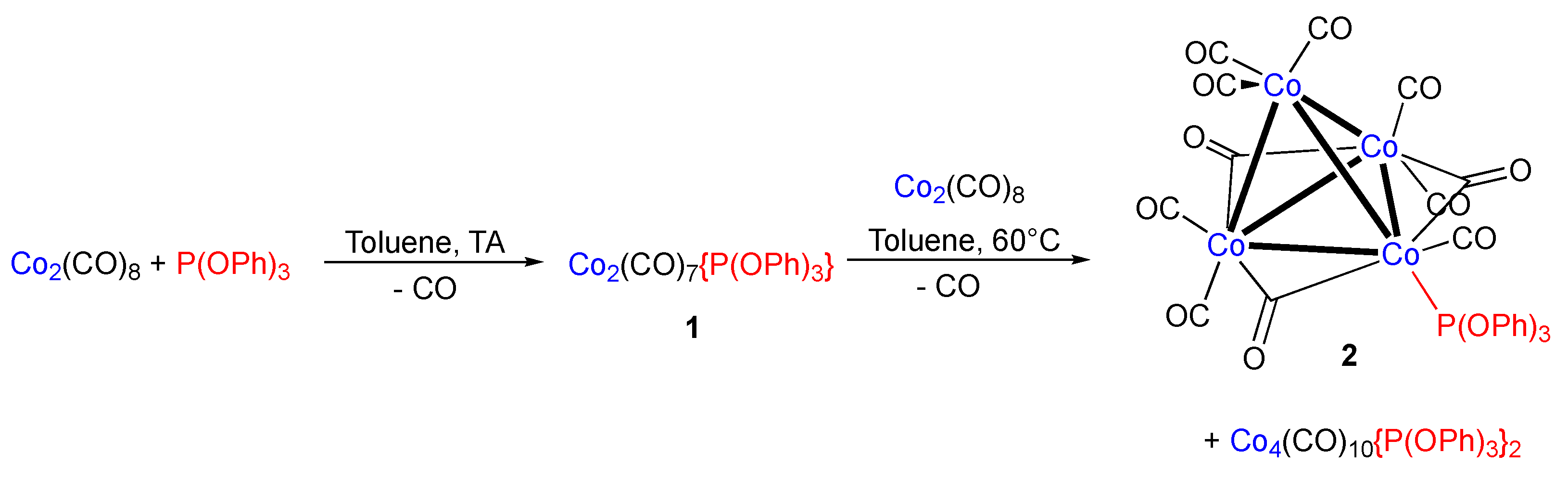
Tris(μ2-carbonyl)octacarbonyl(triphenylphosphite)tetracobalt



Abstract
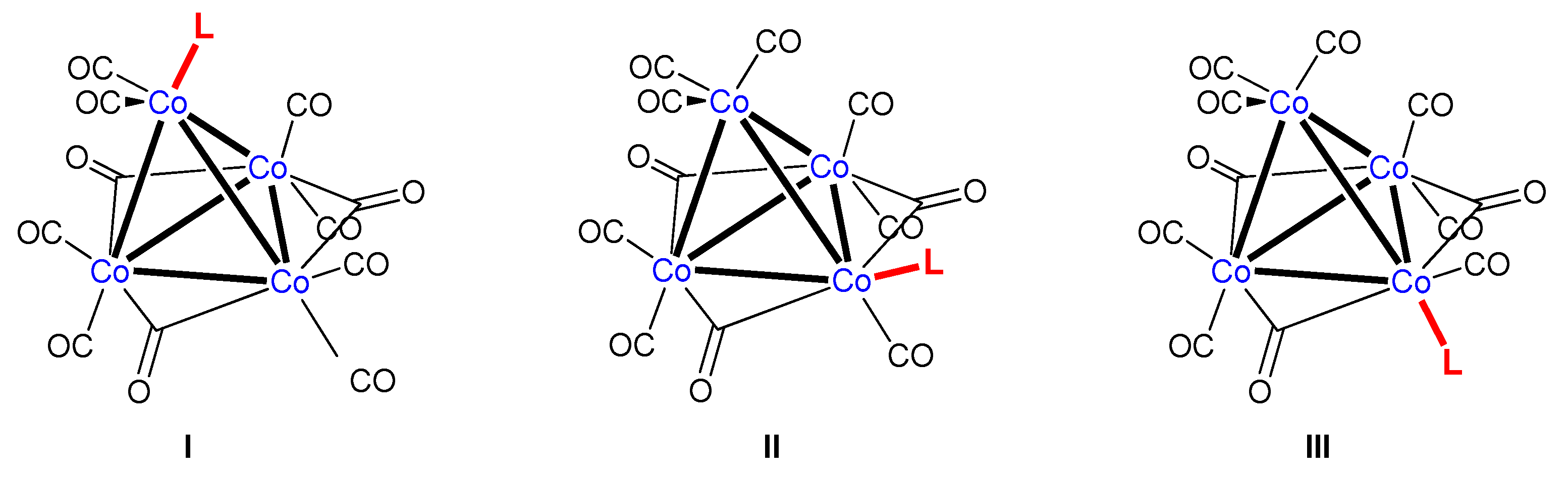
:1. Introduction
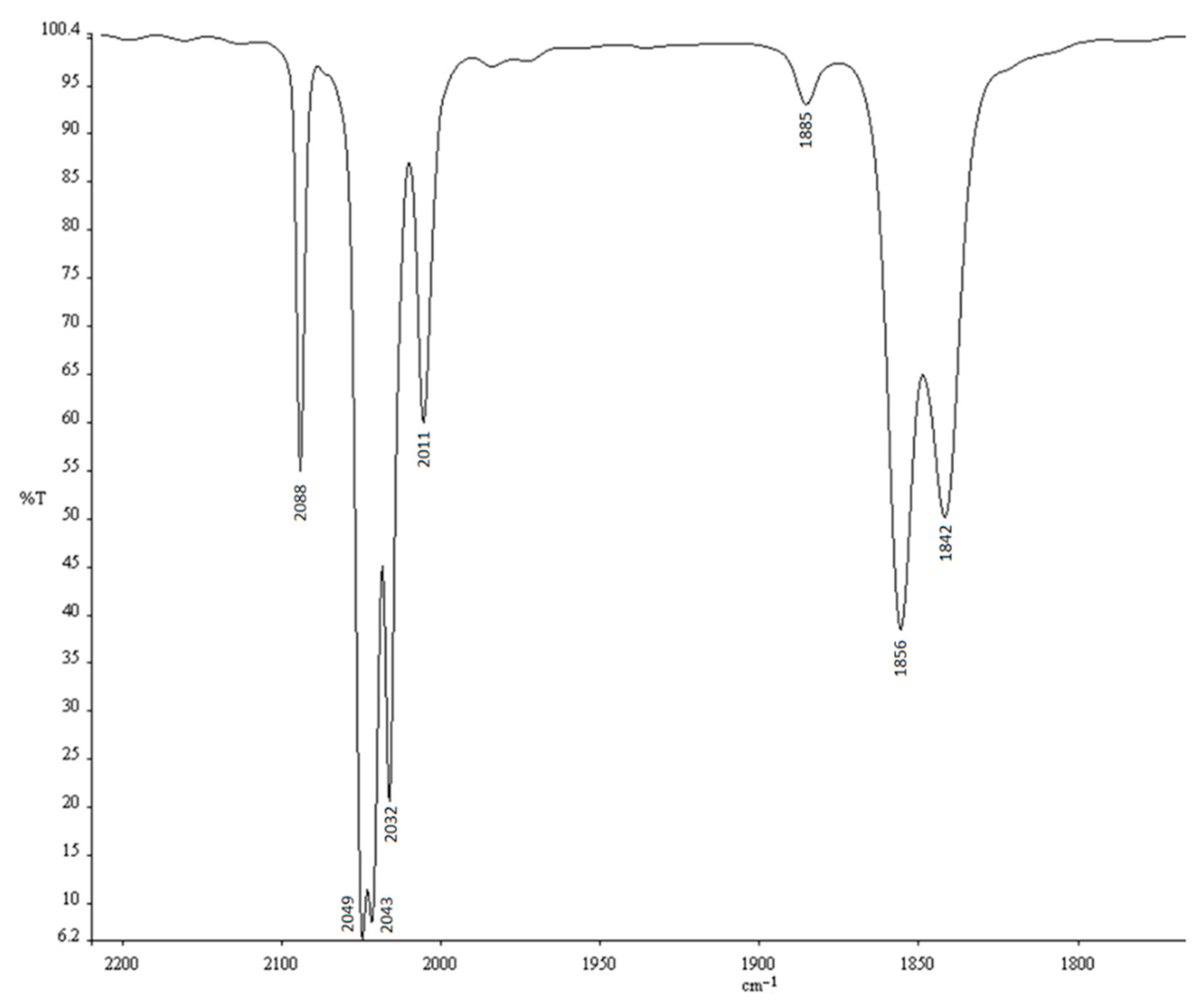
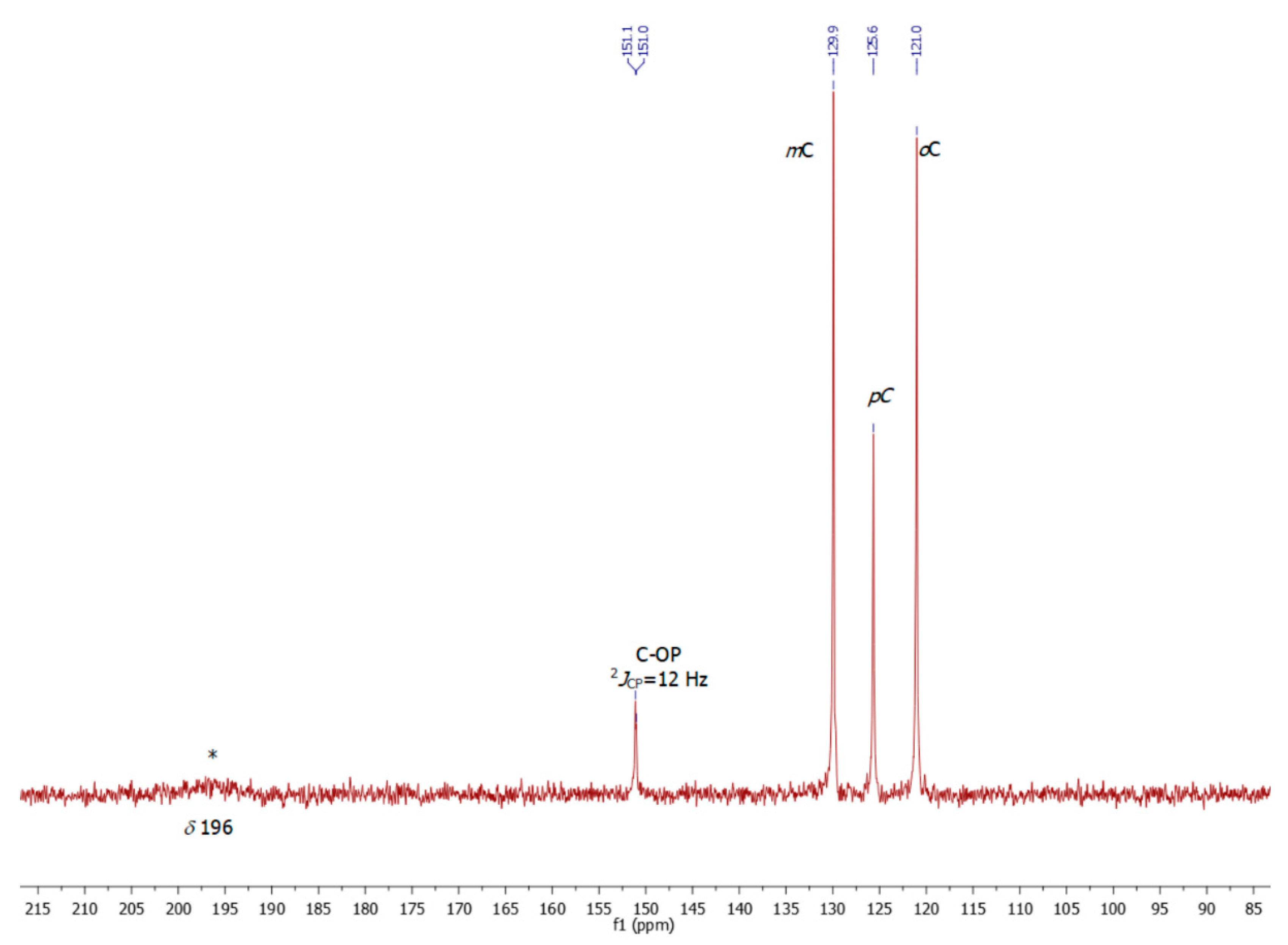
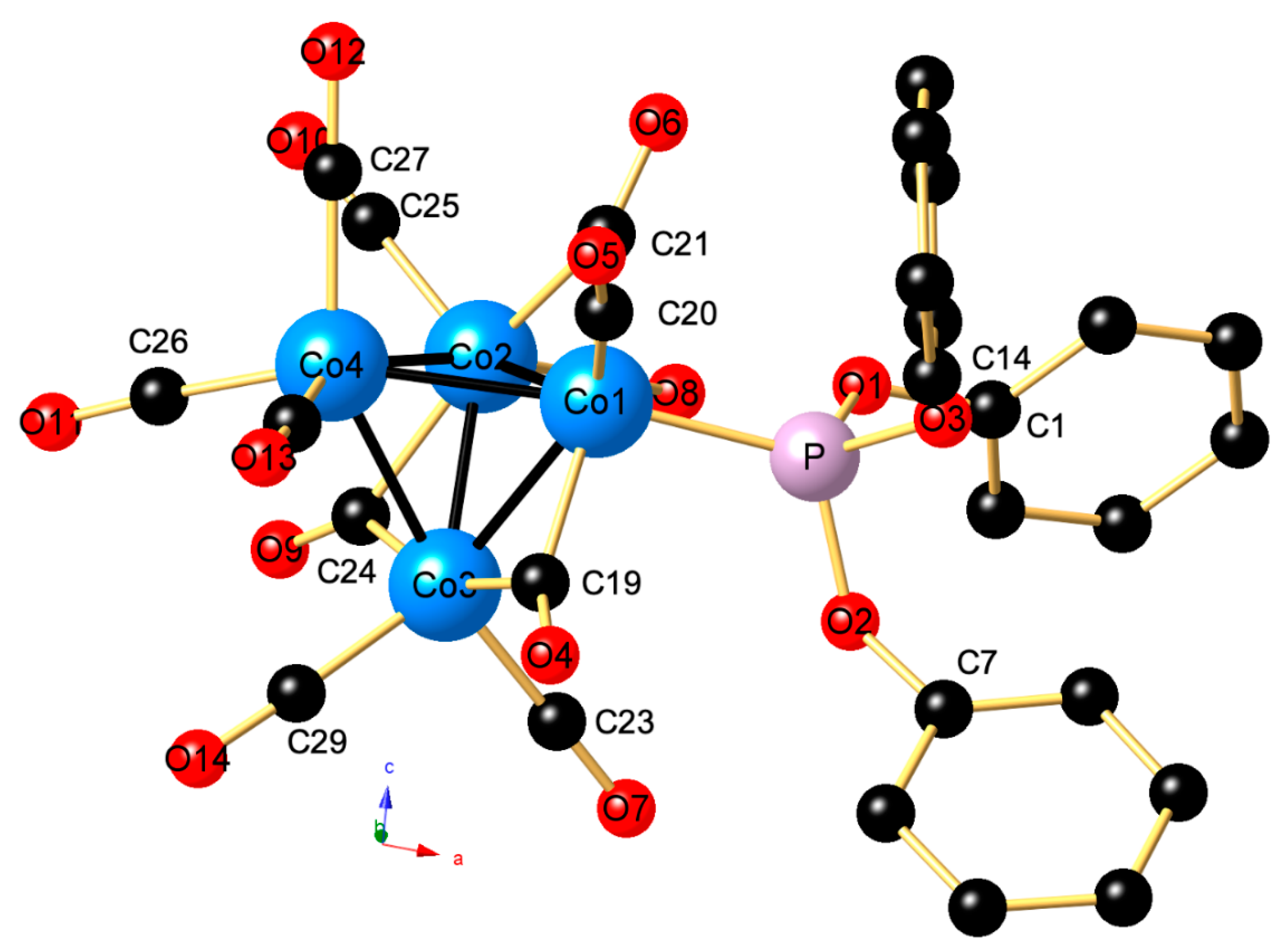
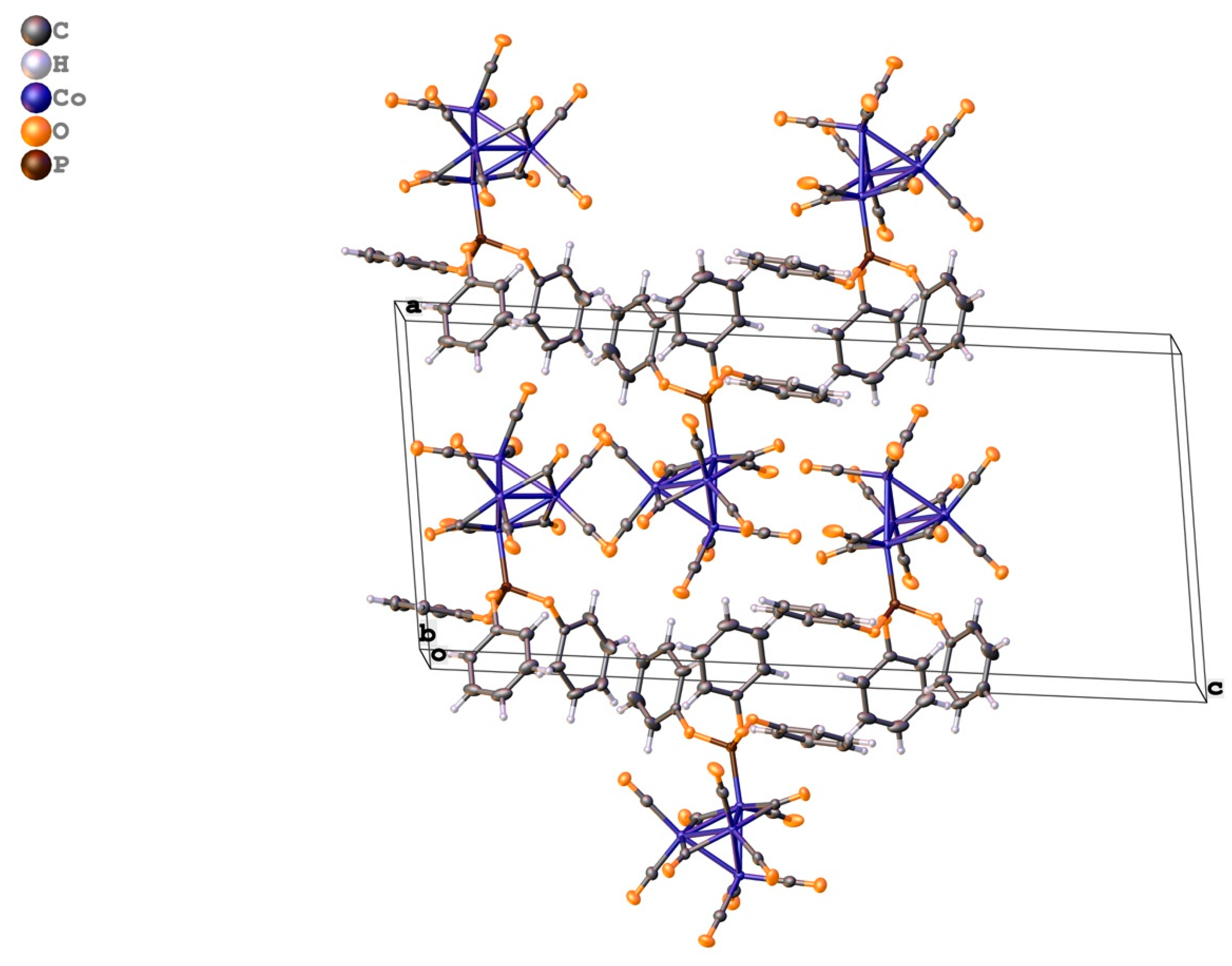
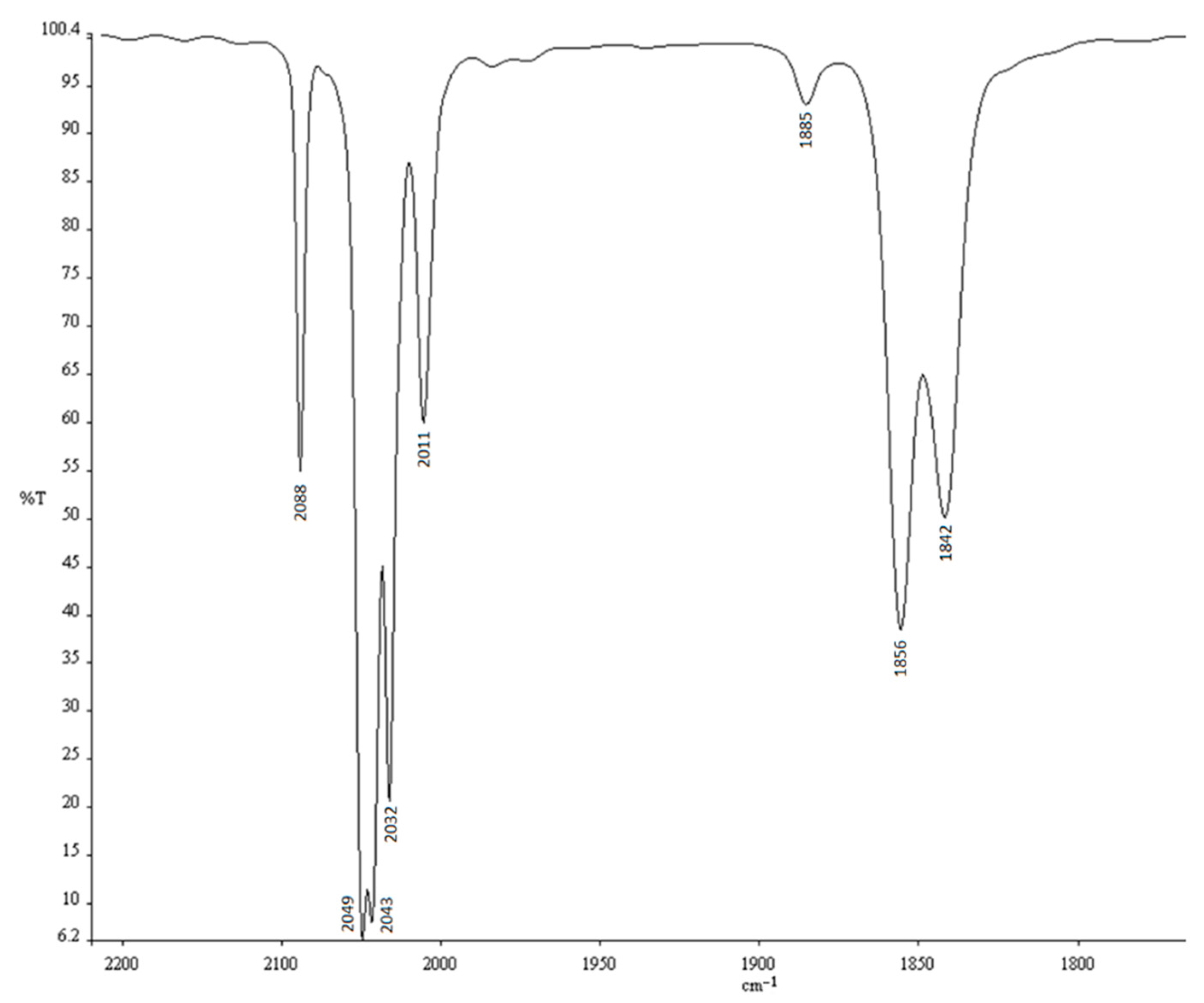
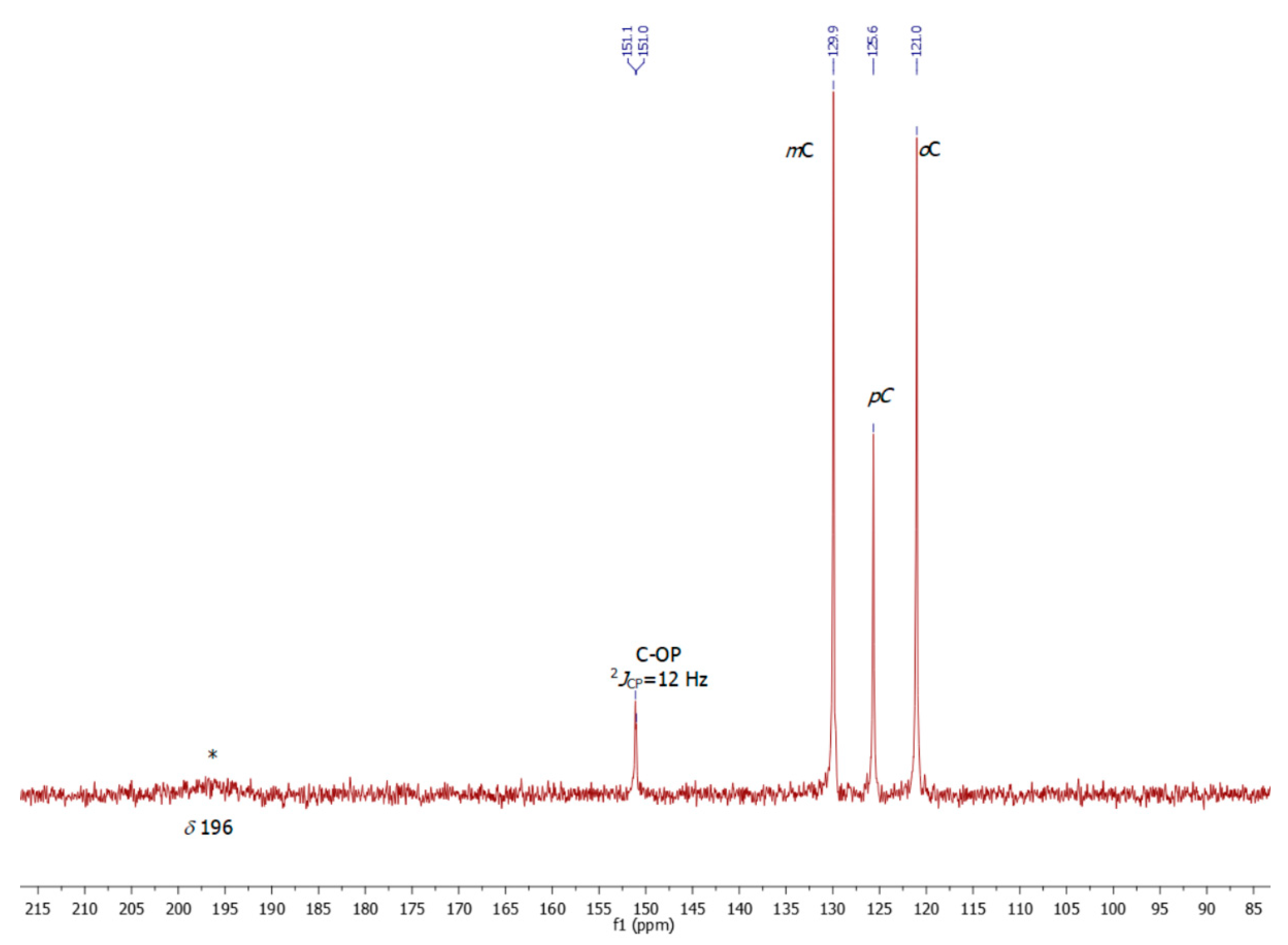
2. Results
3. Discussion
4. Experimental Section
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hieber, W.; Mühlbauer, F.; Ehmann, E.A. Derivate des Kobalt-und Nickelcarbonyls (XVI. Mitteil. über Metallcarbonyle). Ber. Der Dtsch. Chem. Ges. A B Ser. 1932, 65, 1090–1101. [Google Scholar] [CrossRef]
- Chini, P. The Closed Metal Carbonyl Clusters. Inorg. Chim. Acta 1968, 2, 31–51. [Google Scholar] [CrossRef]
- Wei, C.H. Structural Analyses of tetracobalt Dodecarbonyl and tetrarhodium Dodecarbonyl. Crystallographic Treatments of a disordered Structure and a Twinned Composite. Inorg. Chem. 1969, 8, 2384–2397. [Google Scholar] [CrossRef]
- Farrugia, L.J.; Braga, D.; Grepioni, F. A structural redetermination of Co4(CO)12: Evidence for dynamic disorder and the pathway of metal atom migration in the crystalline phase. J. Organomet. Chem. 1999, 573, 60–66. [Google Scholar] [CrossRef]
- Sisak, A.; Sisak, C.; Ungvary, F.; Palyi, G.; Marko, L. Some chemical properties of (arene)nonacarbonylteracobalt complexes and the preparation of (arene)octacarbonyltetracobalt trialkylphosphites. J. Organomet. Chem. 1975, 90, 77–83. [Google Scholar] [CrossRef]
- Bor, G.; Sbrignadello, G.; Marcati, F. Infrared Spectroscopic Studies on Metal Carbonyl Compounds: XV. Assignments of the C-O stretching Infrared Spectra and Force Constants of some (arene)Co4(CO)9 Complexes. J. Organomet. Chem. 1972, 46, 357–368. [Google Scholar] [CrossRef]
- Fliedel, C.; Pattacini, R.; Braunstein, P. Mono-, Di- and Tetranuclear Complexes and Clusters with Bromine-functionalized Bis(diphenylphosphino)amine Ligands. J. Clust. Sci. 2010, 21, 397–415. [Google Scholar] [CrossRef]
- Labroue, D.; Poilblanc, R. Trivalent Phosphorus Derivatives of Cobalt Carbonyls. I. Infrared and NMR Studies of new Substituted Tetracobaltdecacarbonyl Complexes. Inorg. Chim. Acta 1972, 6, 387–390. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. I. Co4(CO)9(μ2-CO)3 derivatives. J. Organomet. Chem. 1979, 171, 89–96. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. 2. Co4(CO)9(μ2-CO)3 derivatives. Inorg. Chem. 1980, 19, 2585–2590. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. 3. Molecular Structures of compound Co4(CO)8(μ-CO)3[P(C6H5)3] and Co4(CO)7(μ-CO)3[P(OCH3)3]2. Inorg. Chem. 1981, 20, 1911–1918. [Google Scholar] [CrossRef]
- Bartl, K.; Boese, R.; Schmid, G. Heteronucleare Clustersysteme XIX. Strukturuntersuchungen an tetraedrischen Carbonylcobalt-clustern; ein Beitrag zur Frage der Bildung von Carbonylbrücken. J. Organomet. Chem. 1981, 206, 331–345. [Google Scholar] [CrossRef]
- Macchi, P.; Garlaschelli, L.; Martinengo, S.; Sironi, A. Charge Density in Transition Metal Clusters: Supported vs Unsupported Metal-Metal Interactions. J. Am. Chem. Soc. 1999, 121, 10428–10429. [Google Scholar] [CrossRef]
- Keller, E.; Vahrenkamp, H. Eisen- und Cobalt-Cluster nach der Propen-Eliminierungs-Methode. Die Cluster Co4(CO)10[P(CH3)2(CH2CHCH2)]2 und Fe2Co(CO)8[μ-SCH3]2[μ-P(CH3)2]. Chem. Ber. 1981, 114, 1111–1123. [Google Scholar] [CrossRef]
- Clément, S.; Guyard, L.; Khatyr, A.; Knorr, M.; Rousselin, Y.; Kubicki, M.M.; Mugnier, Y.; Richeter, S.; Gerbier, P.; Strohmann, C. Synthesis, crystallographic and electrochemical study of ethynyl [2.2] paracyclophane-derived cobalt metallatetrahedranes. J. Organomet. Chem. 2012, 699, 56–66. [Google Scholar] [CrossRef]
- Jourdain, I.; Knorr, M.; Brieger, L.; Strohmann, C. Synthesis of Tris(arylphosphite)-ligated Cobalt(0) Complexes [Co2(CO)6{P(OAr)3}2], and their Reactivity towards Phenylacetylene and Diphenylacetylene. Adv. Chem. Res. 2020, 2. [Google Scholar] [CrossRef]
- Szabo, P.; Fekete, L.; Nagy-Magos, Z.; Marko, L. Phosphorus-containing cobalt carbonyls. III. Monosubstituted derivatives of dicobaltoctacarbonyl with phosphine and phosphites. J. Organomet. Chem. 1968, 12, 245–248. [Google Scholar] [CrossRef]
- Haumann, M.; Meijboom, R.; Moss, J.R.; Roodt, A. Synthesis, crystal structure and hydroformylation activity of triphenylphosphite modified cobalt catalysts. Dalton Trans. 2004, 4, 1679–1686. [Google Scholar] [CrossRef]
- Cohen, M.A.; Kidd, D.R.; Brown, T.L. The structures of Co4(CO)12 and Co4(CO)11[P(OCH3)3] in solution. J. Am. Chem. Soc. 1975, 97, 4408–4409. [Google Scholar] [CrossRef]
- Wade, K. Structural and bonding patterns in cluster chemistry. Adv. Inorg. Chem. Radiochem. 1976, 18, 1–66. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P(OPh)3 | PPh3 | PMe3 | |
|---|---|---|---|
| Co1–Co4 | 2.5037(4) | 2.542(1) | 2.532(2) |
| Co1–Co2 | 2.4937(4) | 2.491(1) | 2.485(2) |
| Co1–Co3 | 2.4568(3) | 2.487(3) | 2.474(2) |
| Co2–Co3 | 2.4494(3) | 2.468(1) | 2.449(2) |
| Co2–Co4 | 2.5153(3) | 2.523(1) | 2.529(2) |
| Co3–Co4 | 2.5129(3) | 2.526(1) | 2.530(2) |
| Average Co–Co | 2.4886 | 2.5062 | 2.4998 |
| Basal-basal | 2.4666 | 2.482 | 2.469 |
| Basal-apical | 2.5106 | 2.530 | 2.530 |
| Co1–P | 2.1546(4) | 2.246(1) | 2.222 |
| Co1-μC19 | 1.9181(12) | 1.908(4) | 1.926 |
| Co3-μC19 | 1.9610(12) | 1.976(4) | 1.989 |
| Co2-μC24 | 1.9365(11) | 1.929(4) | 1.970 |
| Co3-μC24 | 1.9436(12) | 1.951(4) | 1.918 |
| Co1-μC21 | 1.9153(12) | 1.887(5) | 1.852 |
| Co2-μC21 | 1.9712(11) | 1.971(5) | 1.927 |
| Average Co-μC | 1.941 | 1.937 | 1.930 |
| Co1-C20 | 1.7828(13) | 1.758(5) | 1.677 |
| Co2-C22 | 1.8000(13) | 1.794(5) | 1.759 |
| Co2-C25 | 1.7928(13) | 1.778(5) | 1.736 |
| Co3-C23 | 1.8033(12) | 1.789(7) | 1.849 |
| Co3-C29 | 1.7852(12) | 1.776(5) | 1.760 |
| Co4-C26 | 1.8147(12) | 1.832(5) | 1.606 |
| Co4-C27 | 1.8315(13) | 1.827(5) | 1.800 |
| Co4-C28 | 1.8325(14) | 1.822(5) | 1.779 |
| Average Co-C | 1.805 | 1.797 | 1.746 |
| Apical | 1.826 | 1.827 | 1.728 |
| Equatorial | 1.7869 | 1.771 | 1.724 |
| Axial | 1.8017 | 1.792 | 1.804 |
| CSD reference | This work | BAFFET [11] | MSTCOB [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jourdain, I.; Knorr, M.; Strohmann, C.; Scheel, R. Tris(μ2-carbonyl)octacarbonyl(triphenylphosphite)tetracobalt. Molbank 2022, 2022, M1443. https://doi.org/10.3390/M1443
Jourdain I, Knorr M, Strohmann C, Scheel R. Tris(μ2-carbonyl)octacarbonyl(triphenylphosphite)tetracobalt. Molbank. 2022; 2022(3):M1443. https://doi.org/10.3390/M1443
Chicago/Turabian StyleJourdain, Isabelle, Michael Knorr, Carsten Strohmann, and Rebecca Scheel. 2022. "Tris(μ2-carbonyl)octacarbonyl(triphenylphosphite)tetracobalt" Molbank 2022, no. 3: M1443. https://doi.org/10.3390/M1443
APA StyleJourdain, I., Knorr, M., Strohmann, C., & Scheel, R. (2022). Tris(μ2-carbonyl)octacarbonyl(triphenylphosphite)tetracobalt. Molbank, 2022(3), M1443. https://doi.org/10.3390/M1443