
Microwave Assisted Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Some 2-Formimidate-3-carbonitrile Derivatives Bearing 4H-Pyran and Dihydropyridine Moieties



Abstract
:1. Introduction
2. Materials and Methods
2.1. General Procedure for the Conventional Solvothermal Synthesis of 2-Formimidate-3-carbonitrile Derivatives (ii-a to ii-f)
2.2. General Procedure for the Microwave Synthesis of 2-Formimidate-3-carbonitrile Derivatives (ii-a to ii-f)
2.2.1. Ethyl (E)-N-(3-Cyano-4-(2-fluorophenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromen-2-yl)formimidate (ii-a)
2.2.2. Ethyl (E)-N-(4-(Anthracen-9-yl)-3-cyano-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromen-2-yl)formimidate (ii-b)
2.2.3. Ethyl (E)-N-(3-Cyano-4-(2-fluorophenyl)-7,7-dimethyl-5-oxo-1-phenyl-1,4,5,6,7,8-hexahydroquinolin-2-yl)formimidate (ii-c)
2.2.4. Ethyl (E)-N-(3-Cyano-7,7-dimethyl-5-oxo-1,4-diphenyl-1,4,5,6,7,8-hexahydroquinolin-2-yl)formimidate (ii-d)
2.2.5. Ethyl (E)-N-(1-(4-Bromophenyl)-3-cyano-7,7-dimethyl-5-oxo-4-phenyl-1,4,5,6,7,8-hexahydroquinolin-2-yl)formimidate (ii-e)
2.2.6. Ethyl (E)-N-(3-Cyano-7,7-dimethyl-5-oxo-4-phenyl-1-(p-tolyl)-1,4,5,6,7,8-hexahydroquinolin-2-yl)formimidate (ii-f)
2.3. Crystal Structure Determination
2.4. Hirshfeld Surface Analysis
3. Results and Discussion
3.1. Synthesis Consideration and Spectroscopic Characterization
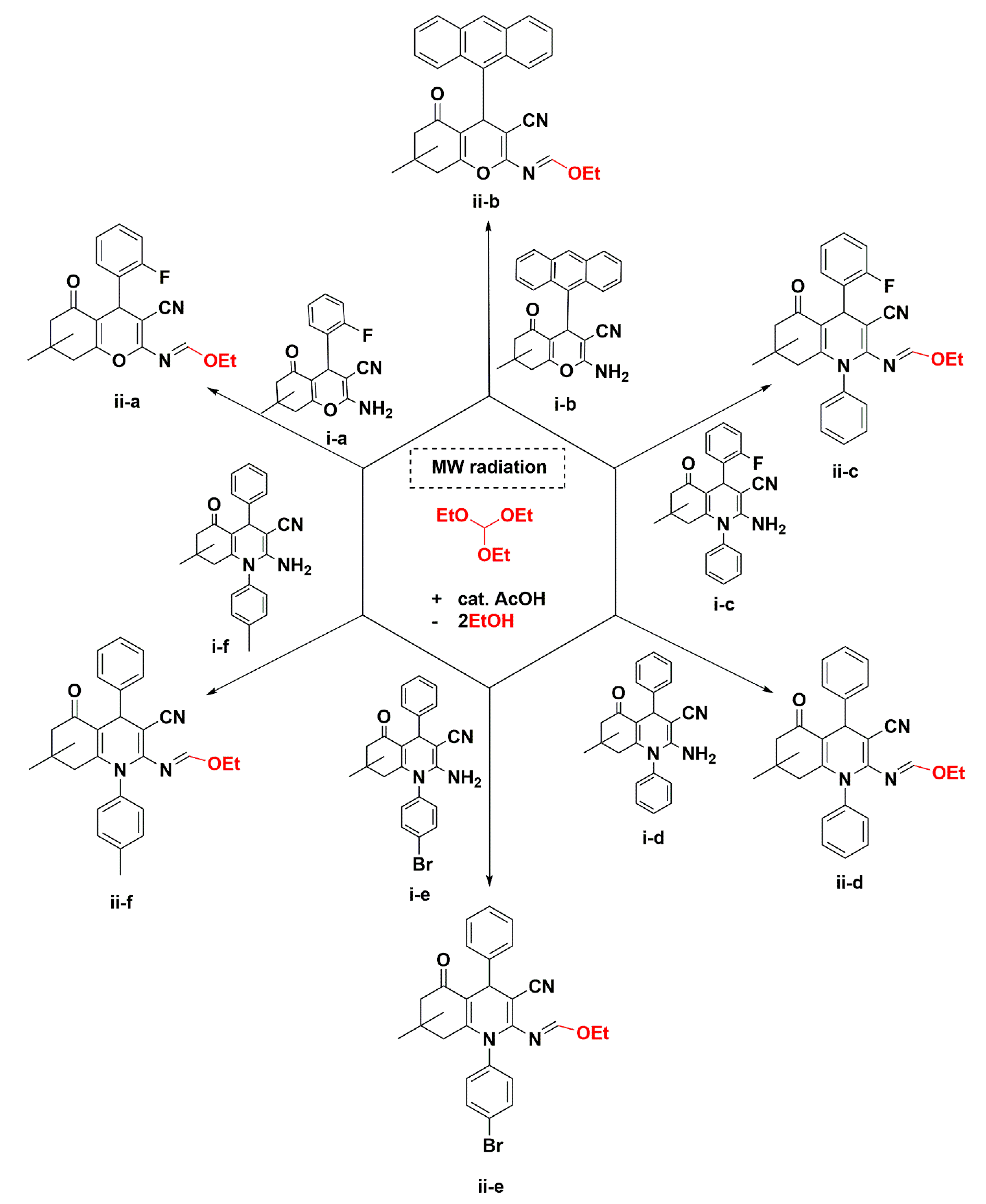
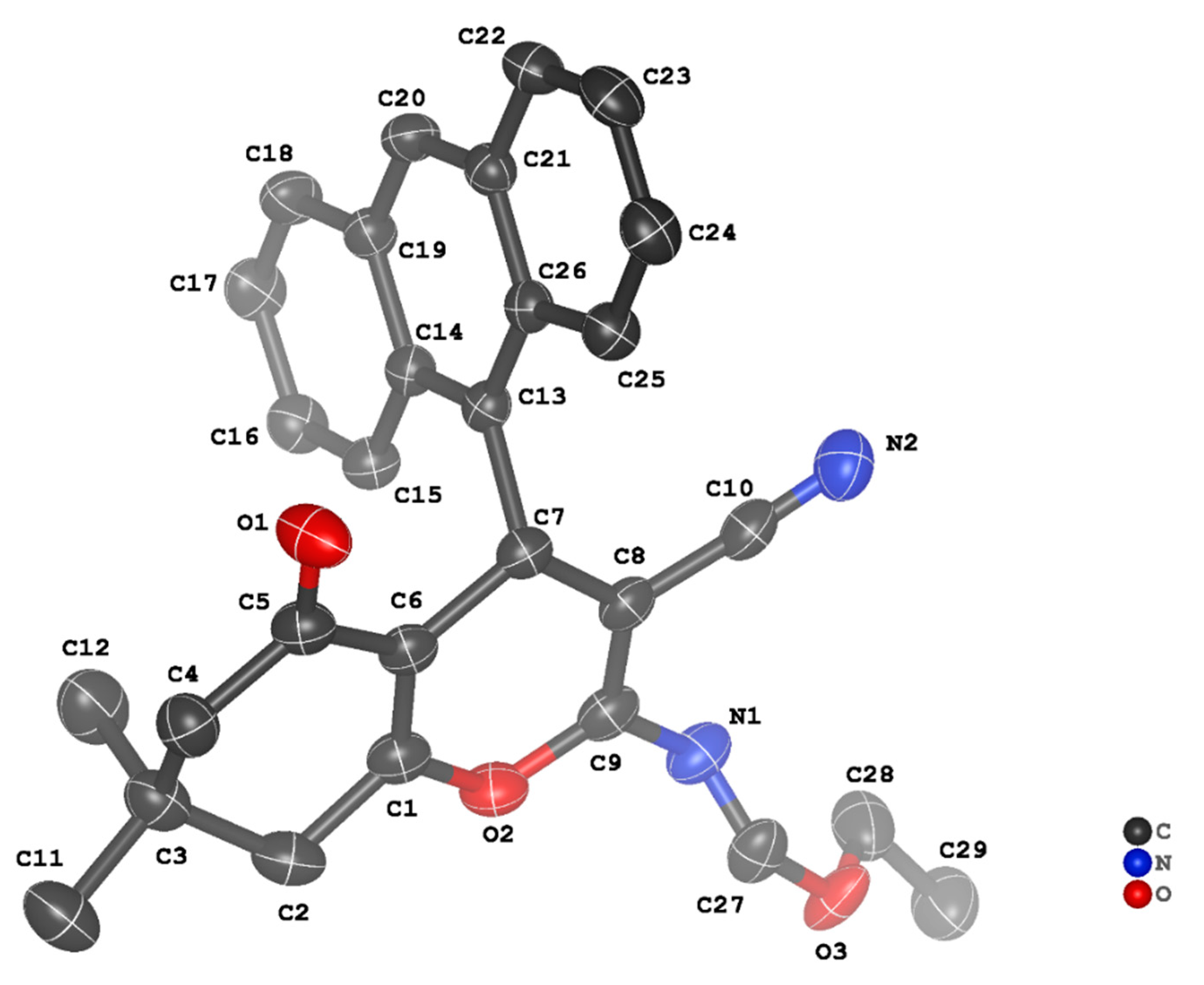
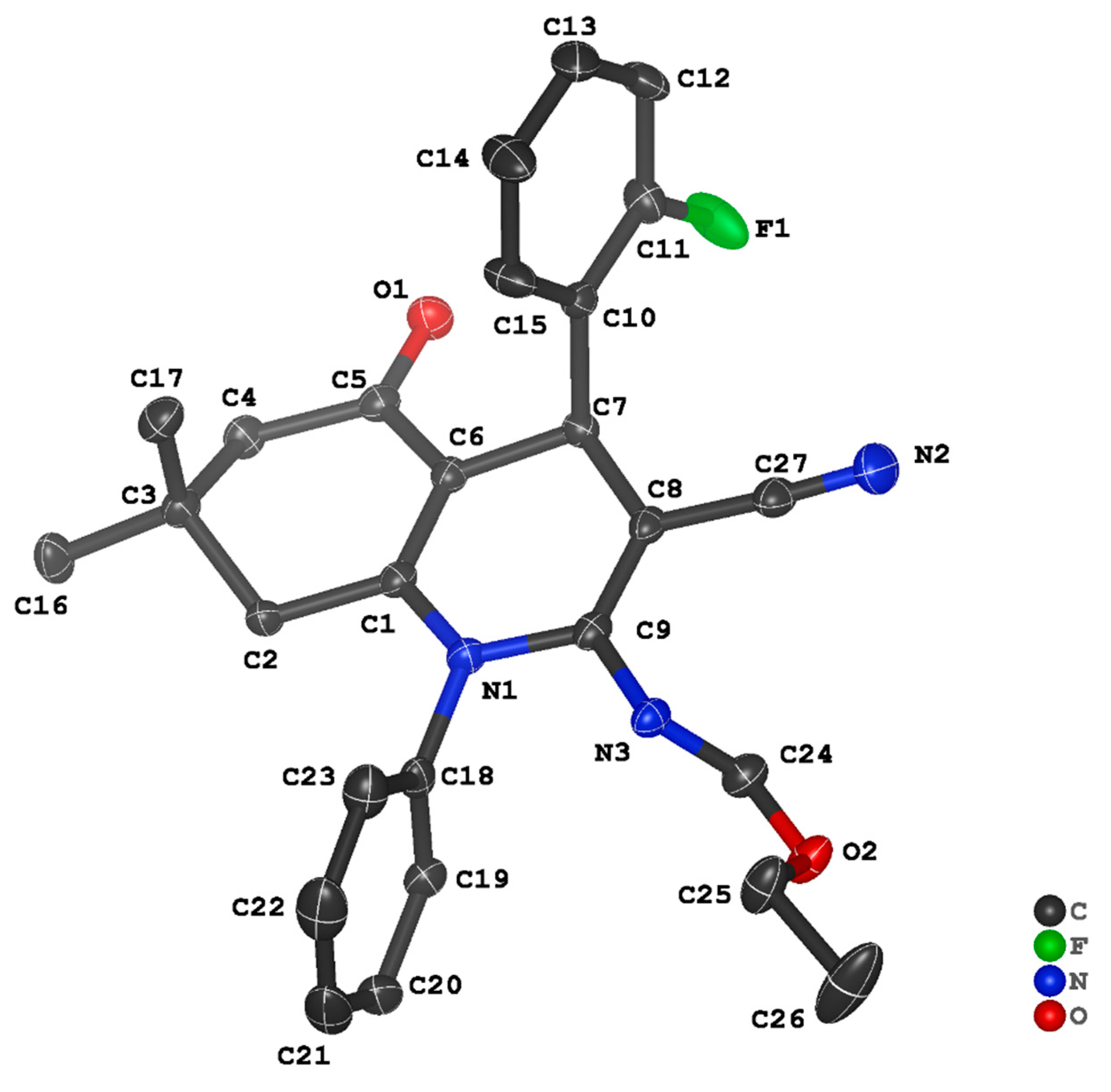
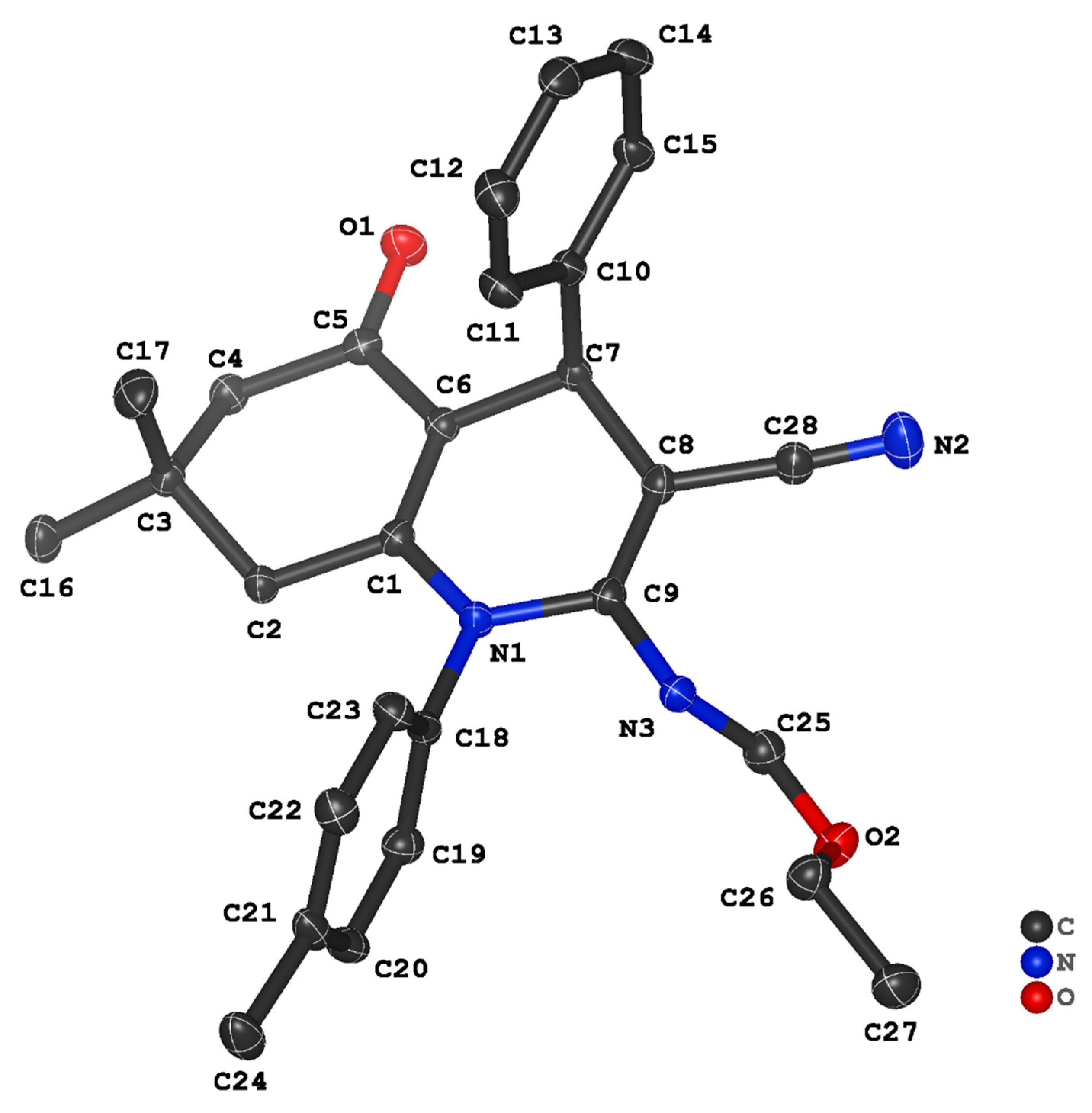
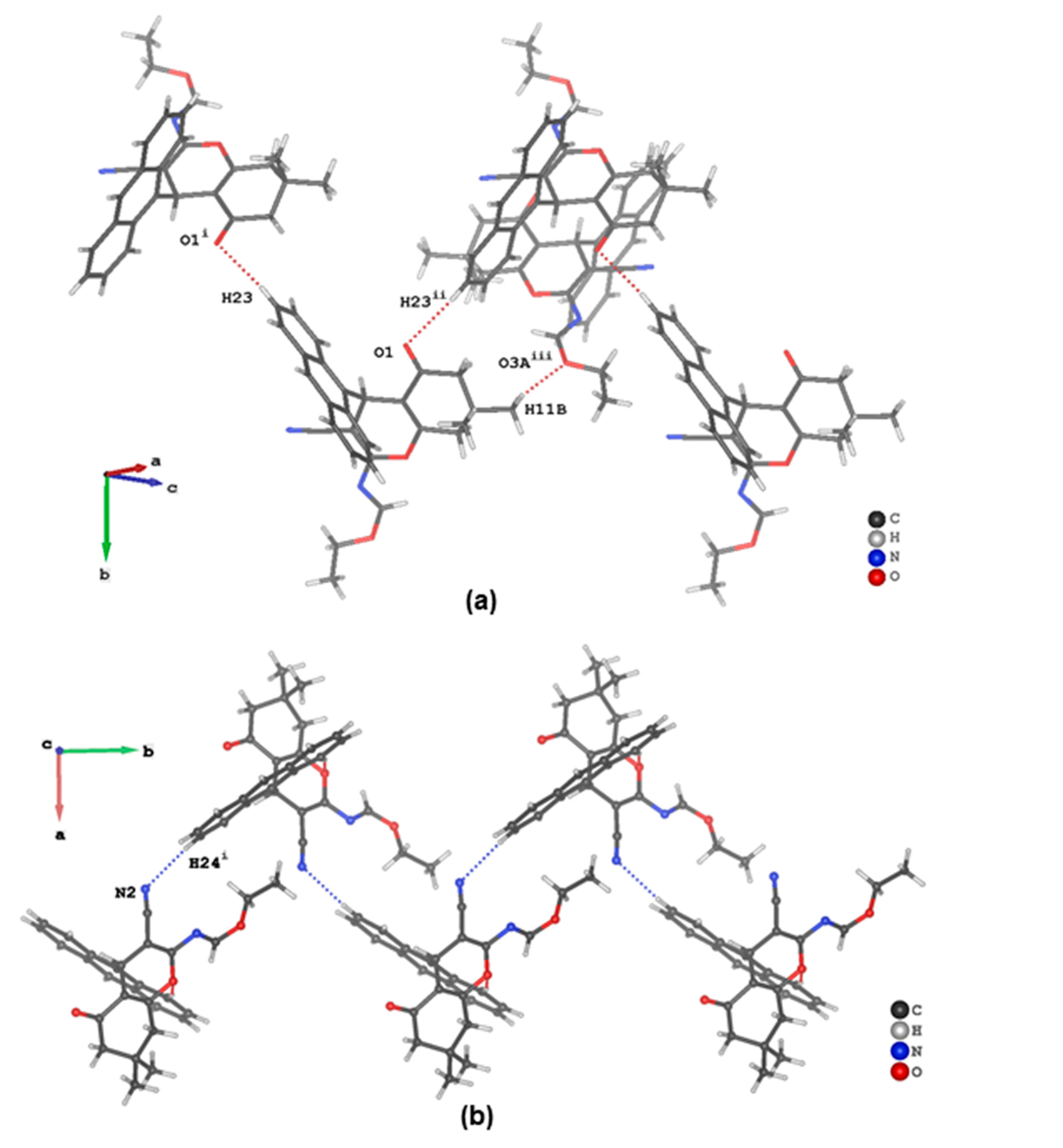
3.2. Crystal Structure Descriptions of ii-b, ii-c and ii-f
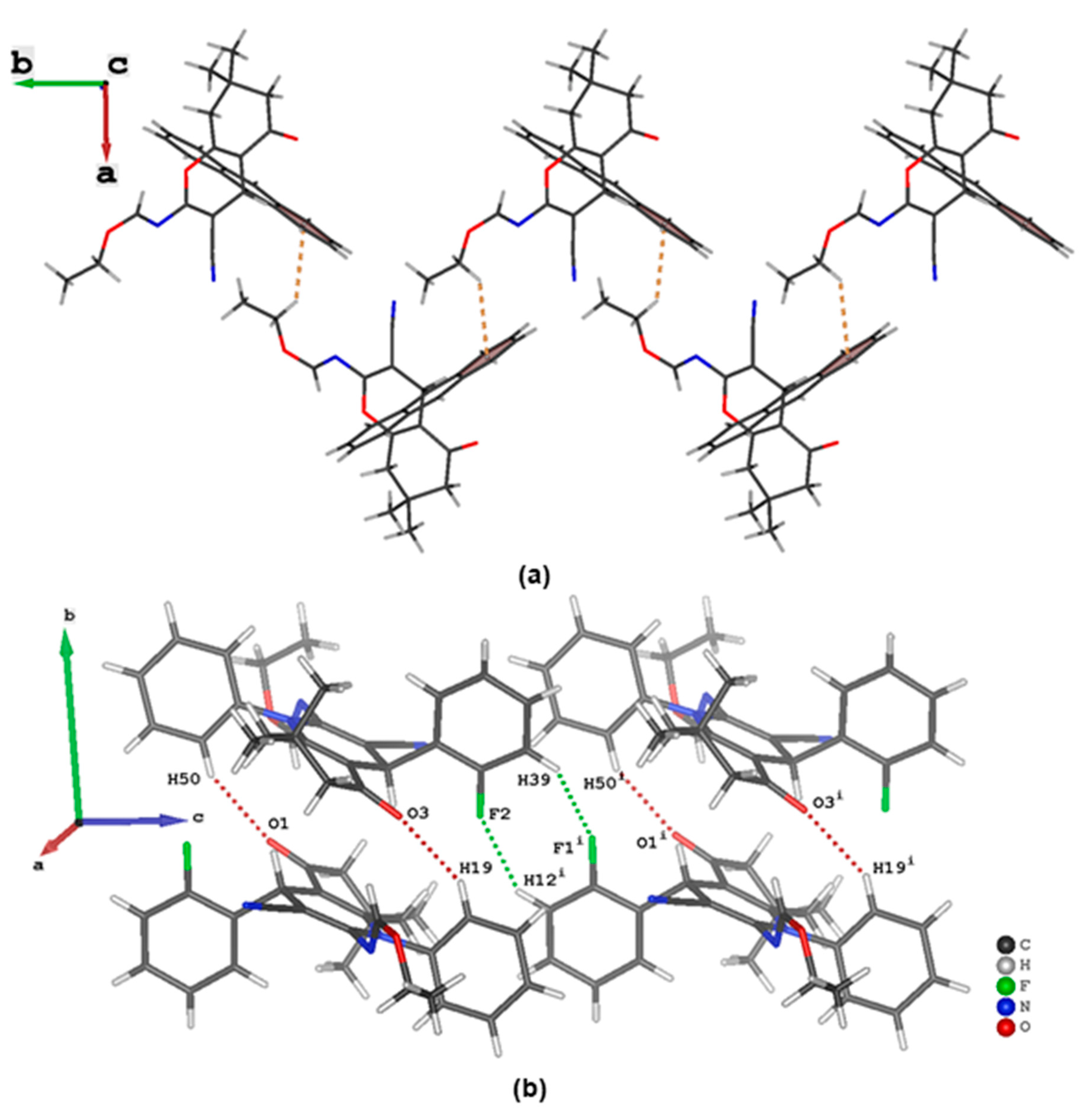
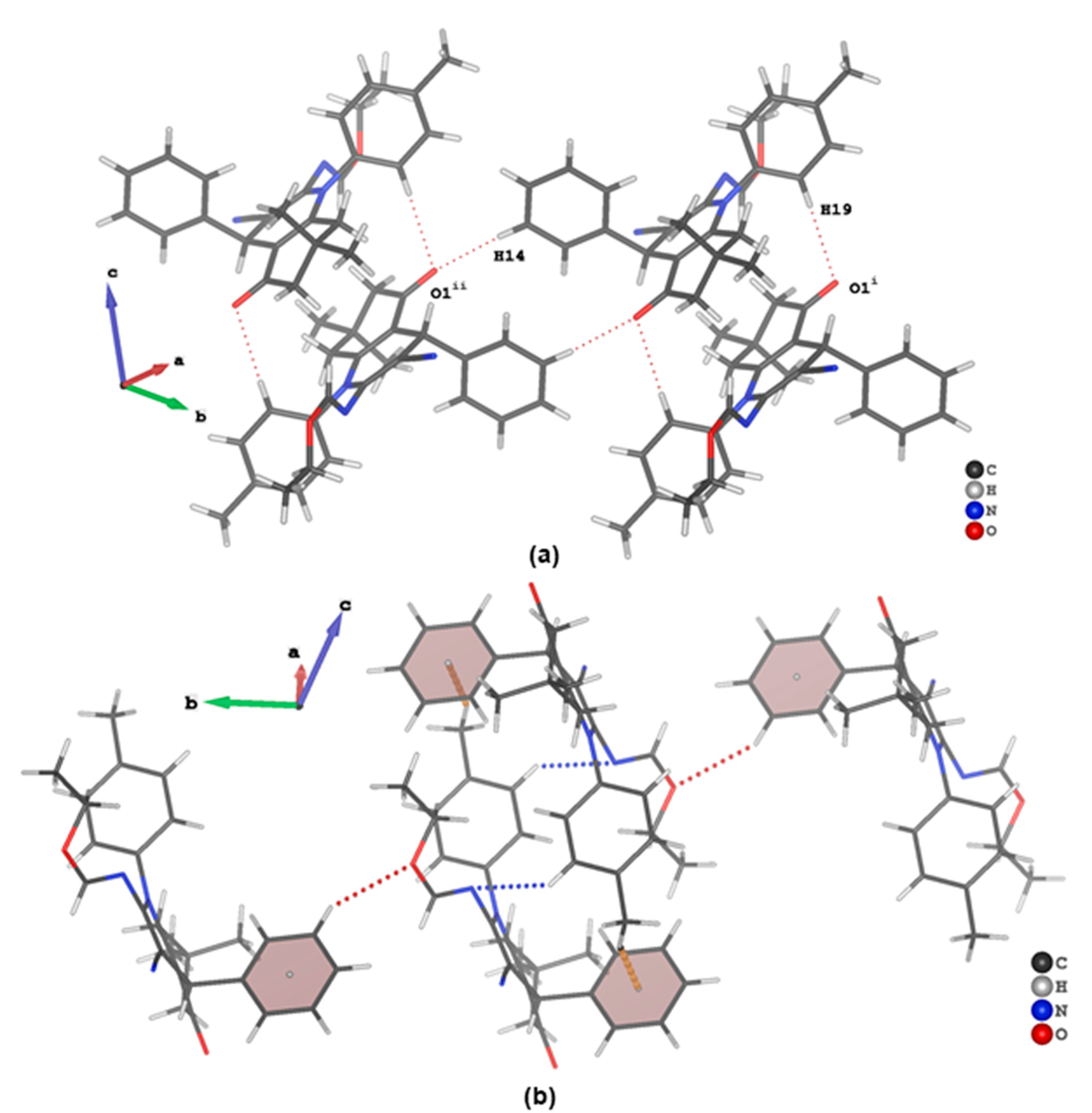
3.3. Evaluation of Intermolecular Interactions in the Crystal Packing of ii-b, ii-c and ii-f
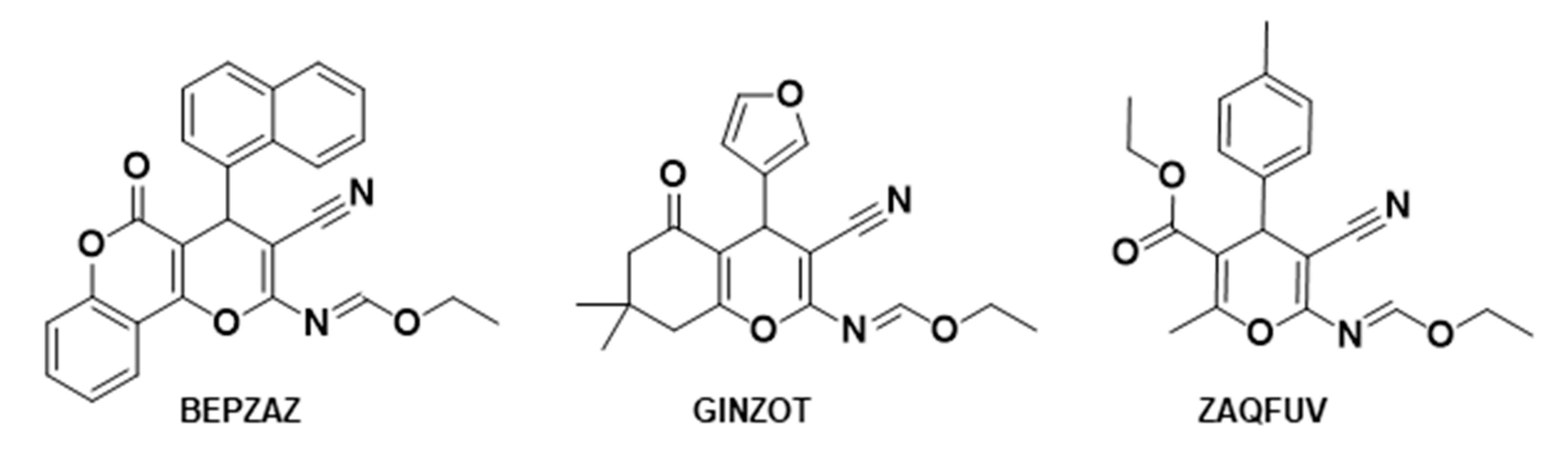
3.4. CSD Survey of Closely Related Compounds
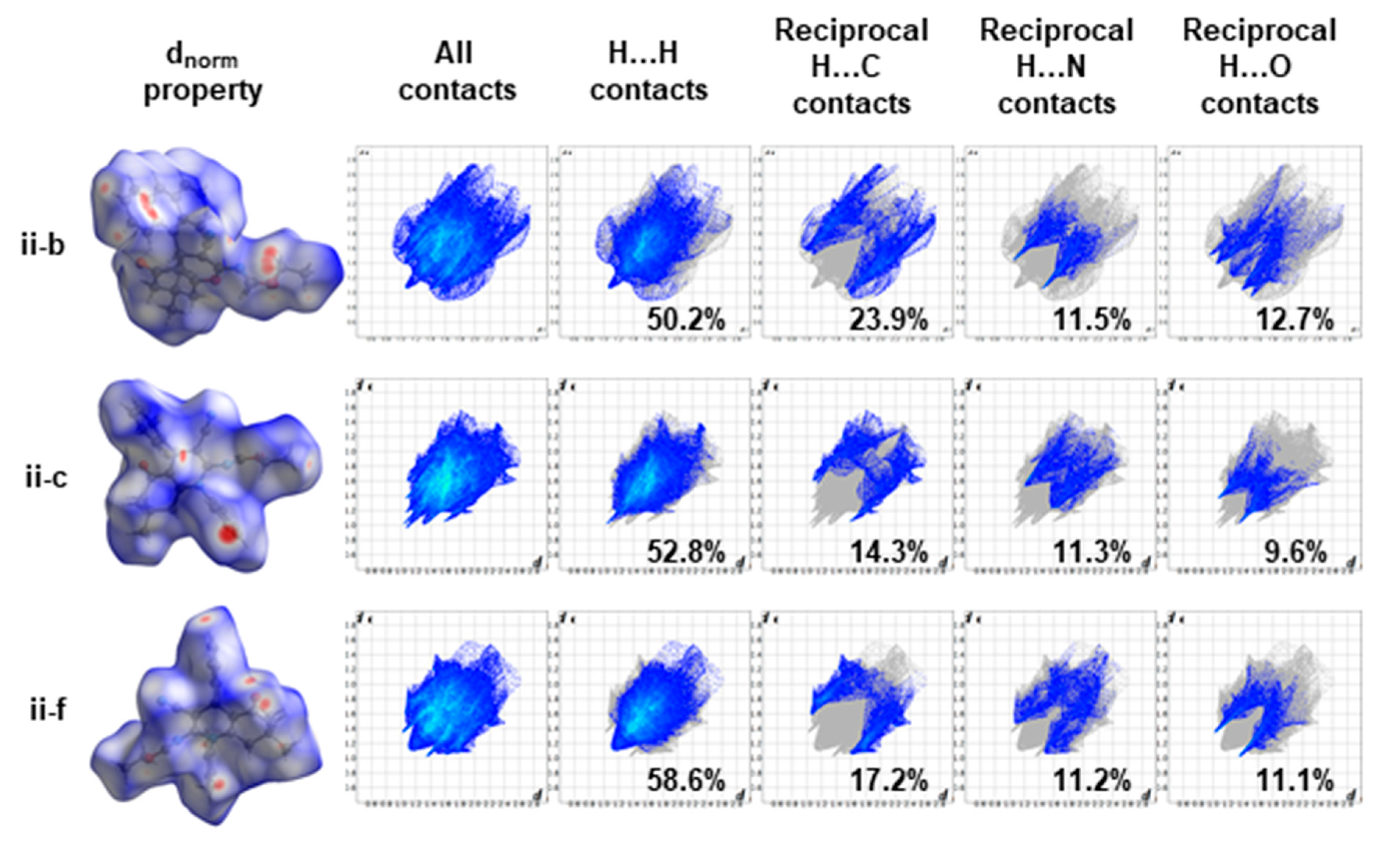
3.5. Hirshfeld Surface Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haggam, R.A.; Assy, M.G.; Mohamed, E.K.; Mohamed, A.S. Synthesis of Pyrano[2,3-d]pyrimidine-2,4-diones and Pyridino[2,3-d]pyrimidine-2,4,6,8-tetraones: Evaluation Antitumor Activity. J. Heterocycl. Chem. 2020, 57, 842–850. [Google Scholar] [CrossRef]
- Suresh, L.; Kumar, P.S.V.; Chandramouli, G.V.P. An efficient one-pot synthesis, characterization and antibacterial activity of novel chromeno-pyrimidine derivatives. J. Mol. Struct. 2017, 1134, 51–58. [Google Scholar] [CrossRef]
- Belhadj, F.; Kibou, Z.; Benabdallah, M.; Aissaoui, M.; Rahmoun, M.N.; Villemin, D.; Choukchou-Braham, N. Synthesis and Biological Evaluation of New Chromenes and Chromeno[2,3-d] pyrimidines. S. Afr. J. Chem. 2021, 75, 150–155. [Google Scholar] [CrossRef]
- El-Sayed, R.; Fadda, A.A. Synthesis of Pharmacological Heterocyclic Derivatives Based Surfactants. J. Oleo Sci. 2016, 65, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu El-Azm, F.S.M.; El-Shahawi, M.M.; Elgubbi, A.S.; Madkour, H.M.F. Design, synthesis, anti-proliferative activity, and molecular docking studies of novel benzo[f]chromene, chromeno [2,3-d]pyrimidines and chromenotriazolo[1,5-c]pyrimidines. Synth. Commun. 2020, 50, 669–683. [Google Scholar] [CrossRef]
- Halawa, A.H.; Elaasser, M.M.; El Kerdawy, A.M.; Abd El-Hady, A.M.A.I.; Emam, H.A.; El-Agrody, A.M. Anticancer activities, molecular docking and structure–activity relationship of novel synthesized 4H-chromene, and 5H-chromeno[2,3-d]pyrimidine candidates. Med. Chem. Res. 2017, 26, 2624–2638. [Google Scholar] [CrossRef]
- Bassyouni, F.; Tarek, M.; Salama, A.; Ibrahim, B.; Salah El Dine, S.; Yassin, N.; Hassanein, A.; Moharam, M.; Abdel-Rehim, M. Promising Antidiabetic and Antimicrobial Agents Based on Fused Pyrimidine Derivatives: Molecular Modeling and Biological Evaluation with Histopathological Effect. Molecules 2021, 26, 2370. [Google Scholar] [CrossRef]
- Gouhar, R.; Abou-Elmagd, W.; El-Zahar, M.; Kamel, M.; El-Ghonamy, D. Synthesis of novel 5,6,7,8,9,10-hexahydropyrimido[4,5-b]quinoline derivatives for antimicrobial and anti-oxidant evaluation. Res. Chem. Intermed. 2017, 43, 1301–1327. [Google Scholar] [CrossRef]
- Taylor, E.C.; Ehrhart, W.A. A Convenient Synthesis of N,N’-Disubstituted Formamidines and Acetamidines1. J. Org. Chem. 1963, 28, 1108–1112. [Google Scholar] [CrossRef]
- Yoon, D.S.; Han, Y.; Stark, T.M.; Haber, J.C.; Gregg, B.T.; Stankovich, S.B. Efficient Synthesis of 4-Aminoquinazoline and Thieno[3,2-d]pyrimidin-4-ylamine Derivatives by Microwave Irradiation. Org. Lett. 2004, 6, 4775–4778. [Google Scholar] [CrossRef]
- Abdelrazek, F.M.; Metz, P.; Kataeva, O.; Jaeger, A.; El-Mahrouky, S.F. Synthesis and molluscicidal activity of new chromene and pyrano[2,3-c]pyrazole derivatives. Arch. Pharm. 2007, 340, 543–548. [Google Scholar] [CrossRef]
- Ameli, S.; Davoodnia, A.; Pordel, M.; Behmadi, H. Synthesis of New Imino Containing Tetrahydrochromeno[2,3-d]pyrimidines. J. Heterocycl. Chem. 2017, 54, 1437–1441. [Google Scholar] [CrossRef]
- Debbabi, M.; Nimbarte, V.D.; Chekir, S.; Chortani, S.; Romdhane, A.; Ben Jannet, H. Design and synthesis of novel potent anticoagulant and anti-tyrosinase pyranopyrimidines and pyranotriazolopyrimidines: Insights from molecular docking and SAR analysis. Bioorganic Chem. 2019, 82, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, M.N.; Huynh, T.H.V.; Abrahamsen, B.; Bastlund, J.F.; Bundgaard, C.; Monrad, O.; Bekker-Jensen, A.; Nielsen, C.W.; Frydenvang, K.; Jensen, A.A.; et al. Structure-Activity Relationship Study of First Selective Inhibitor of Excitatory Amino Acid Transporter Subtype 1: 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101). J. Med. Chem. 2010, 53, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Fadda, A.A.; Youssif, E.H.E. Synthesis of Some New Chromene Derivatives, Part 6. Synth. Commun. 2011, 41, 677–694. [Google Scholar] [CrossRef]
- Hassanien, A.A.; Zahran, M.A.; El-Gaby, M.S.A.; Ghorab, M.M. Utility of 2-amino-4,5,6,8-tetrahydro-7H-chromene-3-carbonitriles in synthesis of chromeno[2,3-d]pyrimidine and chromeno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives of pharmaceutical interest. J. Indian Chem. Soc. 1999, 76, 350–354. [Google Scholar] [CrossRef]
- Hu, J.-L.; Sha, F.; Li, Q.; Wu, X.-Y. Highly enantioselective Michael/cyclization tandem reaction between dimedone and isatylidene malononitriles. Tetrahedron 2018, 74, 7148–7155. [Google Scholar] [CrossRef]
- Kong, X.-X.; Cao, Y.-N.; Xing, Z.; Chen, L.-Z.; Han, G.-F. Synthesis of novel 15-aryl-2,3,4,15-tetrahydrochromeno[2′,3′:4,5]pyrimido[6,1-b]quinazoline-1,9-diones. J. Chem. Res. 2016, 40, 87–91. [Google Scholar] [CrossRef]
- Li, B.; Wang, Z.-X.; Xing, Z.; Chen, L.-Z.; Han, G.-F. Synthesis of novel 2-methyl and 2-cyanomethyl-12-aryl-8,12-dihydro-9H-chromeno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidin-11(10H)-one derivatives. J. Chem. Res. 2015, 39, 30–35. [Google Scholar] [CrossRef]
- Mahdavi, S.M.; Habibi, A.; Dolati, H.; Shahcheragh, S.M.; Sardari, S.; Azerang, P. Synthesis and antimicrobial evaluation of 4H-pyrans and Schiff bases fused 4H-pyran derivatives as inhibitors of Mycobacterium bovis (BCG). Iran. J. Pharm. Res. 2018, 17, 1229–1239. [Google Scholar]
- Mobinikhaledi, A.; Foroughifar, N.; Mosleh, T.; Hamta, A. Synthesis of some novel chromenopyrimidine derivatives and evaluation of their biological activities. Iran. J. Pharm. Res. 2014, 13, 873–879. [Google Scholar] [PubMed]
- Wang, Z.-X.; Li, B.; Xing, Z.; Chen, L.-Z.; Han, G.-F. Synthesis of novel 9,9-dimethyl-8,12-dihydro-9H-chromeno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidin-11(10H)-one derivatives. J. Chem. Res. 2014, 38, 480–485. [Google Scholar] [CrossRef]
- Youssef, M.S.K.; Abeed, A.A.O.; El-Emary, T.I. Synthesis and evaluation of chromene-based compounds containing pyrazole moiety as antimicrobial agents. Heterocycl. Commun. 2017, 23, 55–64. [Google Scholar] [CrossRef]
- Singh, S.K.; Singh, K.N. DBU-catalyzed expeditious and facile multicomponent synthesis of N-arylquinolines under microwave irradiation. Mon. Chem. 2012, 143, 805–808. [Google Scholar] [CrossRef]
- Bruker. APEXII; Bruker AXS: Madison, WI, USA, 2009. [Google Scholar]
- Bruker. SAINT; Bruker AXS: Madison, WI, USA, 2009. [Google Scholar]
- Bruker. SADABS; Bruker AXS: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Commun. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Commun. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.; McKinnon, J.; Wolff, S.; Grimwood, D.; Spackman, P.; Jayatilaka, D.; Spackman, M. CrystalExplorer17; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der Waals volumes and radii of metals in covalent compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Zanin, L.L.; Jimenez, D.E.Q.; de Jesus, M.P.; Diniz, L.F.; Ellena, J.; Porto, A.L.M. Synthesis and X-ray crystal structures of polyfunctionalized 4H-chromene derivatives via tricomponent reaction with Knoevenagel adducts as intermediates in aqueous medium. J. Mol. Struct. 2021, 1223, 129226. [Google Scholar] [CrossRef]
- Ramesh, R.; Maheswari, S.; Malecki, J.G.; Lalitha, A. NaN3 Catalyzed Highly Convenient Access to Functionalized 4H-chromenes: A Green One-pot Approach for Diversity Amplification. Polycycl. Aromat. Compd. 2020, 40, 1581–1594. [Google Scholar] [CrossRef]
- Maharramov, A.; Kaya, R.; Taslimi, P.; Kurbanova, M.; Sadigova, A.; Farzaliyev, V.; Sujayev, A.; Gulçin, İ. Synthesis, crystal structure, and biological evaluation of optically active 2-amino-4-aryl-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromen-3-carbonitriles: Antiepileptic, antidiabetic, and anticholinergics potentials. Arch. Pharm. 2019, 352, 1800317. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, X.-S.; Zhang, M.-M.; Li, Y.-L.; Shi, D.-Q. 2-Amino-4-(2-chlorophenyl)-7,7-dimethyl-1-(4-methylphenyl)-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carbonitrile. Acta Crystallogr. Sect. E 2006, 62, o1184–o1186. [Google Scholar] [CrossRef]
- Al-Masoudi, N.A.; Mohammed, H.H.; Hamdy, A.M.; Akrawi, O.A.; Eleya, N.; Spannenberg, A.; Pannecouque, C.; Langer, P. Synthesis and anti-HIV Activity of New Fused Chromene Derivatives Derived from 2-Amino-4-(1-naphthyl)-5-oxo-4H,5H-pyrano[3,2- c]chromene-3-carbonitrile. Z. Nat. B 2013, 68, 229–238. [Google Scholar] [CrossRef]
- Shi, Q.-Z.; Cao, Y.-N.; Ma, S.-B.; Wang, G.-X.; Han, G.-F.; Xing, Z. Synthesis of Novel Ethyl 1-aryl-3-methyl-8-oxo-1,8-dihydropyrano[2′,3′:4,5] Pyrimido[6,1-b]Quinazoline-2-carboxylate Derivatives. J. Chem. Res. 2016, 40, 767–771. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ii-b | ii-c | ii-f |
|---|---|---|---|
| Empirical formula | C29H26N2O3 | C27H26FN3O2 | C28H29N3O2 |
| Formula weight | 450.52 | 443.51 | 439.54 |
| Temperature/K | 150 | 100 | 99.99 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21 | P-1 |
| a/Å | 12.811(3) | 9.1077(8) | 9.6966(2) |
| b/Å | 13.560(2) | 24.039(2) | 10.2417(2) |
| c/Å | 14.965(3) | 11.0293(10) | 12.4853(2) |
| α/° | 90 | 90 | 103.850(1) |
| β/° | 115.110(2) | 105.3140(10) | 90.328(1) |
| γ/° | 90 | 90 | 102.621(1) |
| Volume/Å3 | 2354.0(8) | 2329.0(4) | 1172.57(4) |
| Z | 4 | 4 | 2 |
| ρcalcg/cm3 | 1.271 | 1.265 | 1.245 |
| μ/mm−1 | 0.083 | 0.086 | 0.079 |
| F(000) | 952.0 | 936.0 | 468.0 |
| Crystal size/mm3 | 0.24 × 0.16 × 0.11 | 0.34 × 0.26 × 0.21 | 0.23 × 0.18 × 0.14 |
| 2Θ range for data collection/° | 4.25 to 52.256 | 3.388 to 54.268 | 4.206 to 56.9 |
| Index ranges | −15 ≤ h ≤ 15 −16 ≤ k ≤ 15 −18 ≤ l ≤ 14 | −11 ≤ h ≤ 10 −30 ≤ k ≤ 30 −14 ≤ l ≤ 14 | −12 ≤ h ≤ 12 −13 ≤ k ≤ 13 −16 ≤ l ≤ 14 |
| Reflections collected | 17738 | 34566 | 23267 |
| Independent reflections | 4523 Rint = 0.0230 Rsigma = 0.0225 | 10,085 Rint = 0.0166 Rsigma = 0.0162 | 5772 Rint = 0.0262 Rsigma = 0.0269 |
| Data/restraints/parameters | 4523/0/328 | 10085/21/610 | 5772/0/302 |
| Goodness-of-fit on F2 | 1.022 | 1.030 | 1.030 |
| Final R indexes [I>=2σ (I)] | R1 = 0.0392 wR2 = 0.0950 | R1 = 0.0323 wR2 = 0.0844 | R1 = 0.0432 wR2 = 0.1090 |
| Final R indexes [all data] | R1 = 0.0563 wR2 = 0.1059 | R1 = 0.0346 wR2 = 0.0865 | R1 = 0.0570 wR2 = 0.1171 |
| Largest diff. peak/hole/e Å−3 | 0.26/−0.16 | 0.38/−0.18 | 0.36/−0.27 |
| Flack parameter | - | 0.07(14) | - |
| D | H | A | d(D-H)/Å | d(H…A)/Å | d(D…A)/Å | D-H…A/° |
|---|---|---|---|---|---|---|
| Compound ii-b | ||||||
| C11 | H11C | O3A i | 0.98 | 2.66 | 3.481(3) | 142 |
| C15 | H15 | O2 | 0.95 | 2.62 | 3.559(2) | 168 |
| C23 | H23 | O1 ii | 0.95 | 2.57 | 3.487(2) | 164 |
| C24 | H24 | N2 iii | 0.95 | 2.63 | 3.574(2) | 170 |
| C28 | H28A | πanthracenyl iv | 0.98 | 2.92 | 3.692(2) | 136 |
| Compound ii-c | ||||||
| C12 | H12 | F2 i | 0.95 | 2.58 | 3.361(3) | 140 |
| C19 | H19 | O3 | 0.95 | 2.56 | 3.396(3) | 147 |
| C50 | H50 | O1 | 0.95 | 2.52 | 3.371(3) | 149 |
| Compound ii-f | ||||||
| C19 | H19 | O1 i | 0.95 | 2.52 | 3.440(2) | 163 |
| C12 | H12 | O2 ii | 0.95 | 2.65 | 3.469(2) | 145 |
| C14 | H14 | O1 iii | 0.95 | 2.55 | 3.484(2) | 170 |
| C22 | H22 | N3 iv | 0.95 | 2.66 | 3.351(2) | 131 |
| C24 | H24C | πphenyl iv | 0.98 | 2.66 | 3.558(2) | 152 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamisa, S.J.; Omondi, B. Microwave Assisted Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Some 2-Formimidate-3-carbonitrile Derivatives Bearing 4H-Pyran and Dihydropyridine Moieties. Molbank 2022, 2022, M1364. https://doi.org/10.3390/M1364
Zamisa SJ, Omondi B. Microwave Assisted Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Some 2-Formimidate-3-carbonitrile Derivatives Bearing 4H-Pyran and Dihydropyridine Moieties. Molbank. 2022; 2022(2):M1364. https://doi.org/10.3390/M1364
Chicago/Turabian StyleZamisa, Sizwe J., and Bernard Omondi. 2022. "Microwave Assisted Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Some 2-Formimidate-3-carbonitrile Derivatives Bearing 4H-Pyran and Dihydropyridine Moieties" Molbank 2022, no. 2: M1364. https://doi.org/10.3390/M1364
APA StyleZamisa, S. J., & Omondi, B. (2022). Microwave Assisted Synthesis, Crystal Structure and Hirshfeld Surface Analysis of Some 2-Formimidate-3-carbonitrile Derivatives Bearing 4H-Pyran and Dihydropyridine Moieties. Molbank, 2022(2), M1364. https://doi.org/10.3390/M1364