2. Results
A concentrated solution of aniline in acetic acid (in demonstration, 44 wt% strength with 4.65 g of aniline was used) reacts vigorously and alongside substantial exotherm with neat disulfur dichloride. This is allowed to react with external cooling (room-temperature water bath) for 30 min and then heated at 80 °C for 4 h. After this time, the reaction forms a thick slurry of brown precipitate. To aid in the collection of the target solid, toluene (50 mL) is added. The filtrate contains a variety of impurities; some major components identified were 4-chloroaniline and
N-oxidised species (e.g., azobenzene). These are removed by washing the precipitate with toluene (50 mL), followed by DCM (50 mL). The precipitate contains the title compound, although it also contains elemental sulfur and small amounts of other organic impurities. Often, crystalline forms of sulfur are collected, although the title compound is entirely amorphous (upon inspection under microscope), as per previous literature reports [
13]. To form crystalline polymorphs, an ion-exchange can be performed with a sufficiently large counterion source (e.g., SbCl
6), followed by slow-growth solution-recrystallisation from polar-organic solvents [
14].
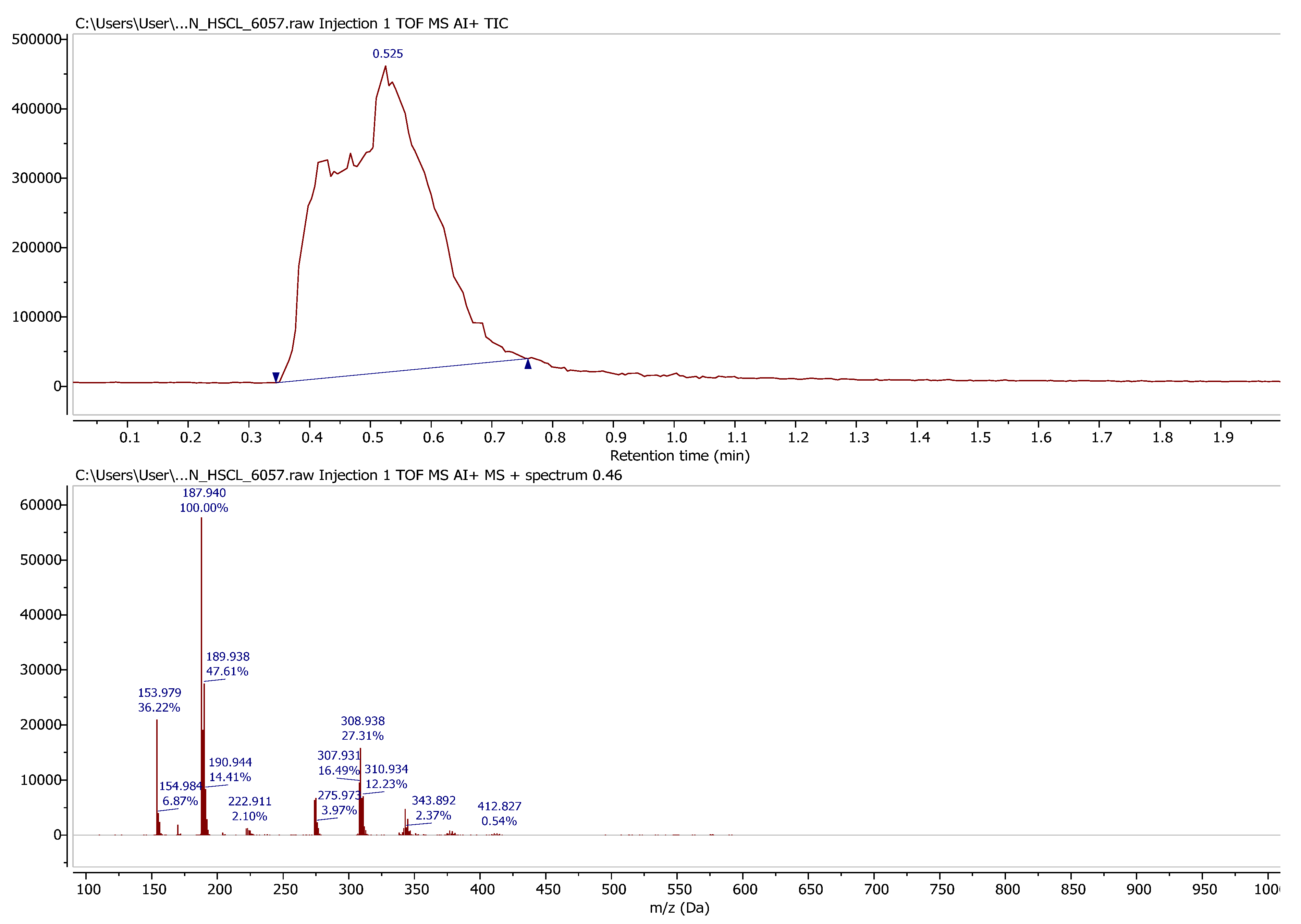
While generally challenging to characterise due to its lack of solubility, the solid product can be analysed by solid-phase ASAP-MS (
Figure 1). The mass peak observed corresponds to the mass of the anticipated Herz cation, and the isotope pattern is also consistent with the molecular formula C
6H
3S
2NCl.
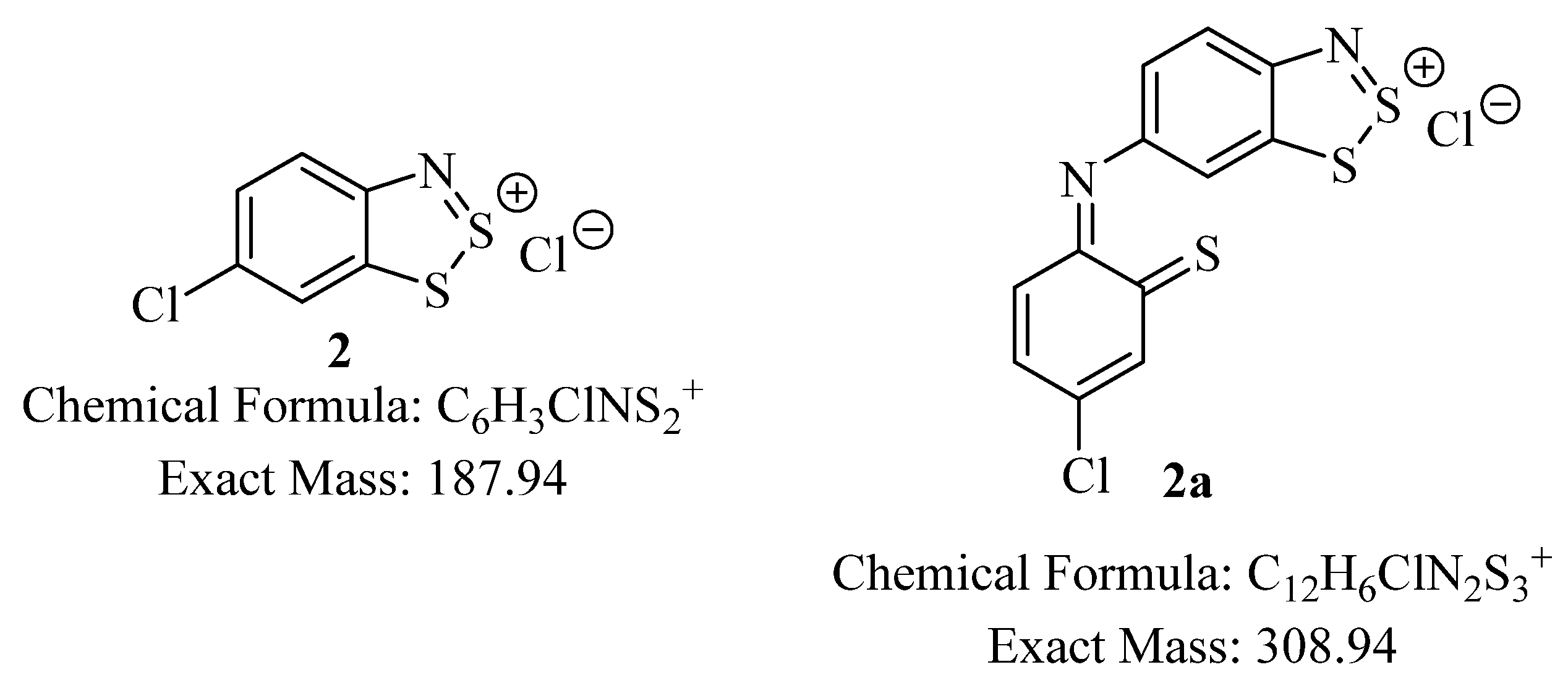
A minor impurity with a Cl-containing splitting pattern is detected with mass 308.938 Da; this is consistent with a molecular formula of C
12H
6ClN
2S
3. This impurity (like the titled compound) must be insoluble in organic solvents and could be the result of two units of (
2) coupling together, with ring-opening and a nucleophilic displacement of the 4-Cl (
2a,
Figure 2), although there is no additional supporting evidence of this structure. After accounting for the elemental sulfur contamination, the yield of target compound
2 was 10.8 g (61% isolated yield).
3. Discussion
At a small scale, the dropwise addition of disulfur dichloride to the aniline solution and with the use of an external cooling bath (room-temperature water) is sufficient to control the reaction. Following further heating, the reaction time is required to complete the process. Different sources have suggested various figures, although in demonstration the optimal heating was found to be 80 °C for four hours, but only after an initial 30 min of stirring with the aforementioned external cooling in order to prevent thermal runaway.
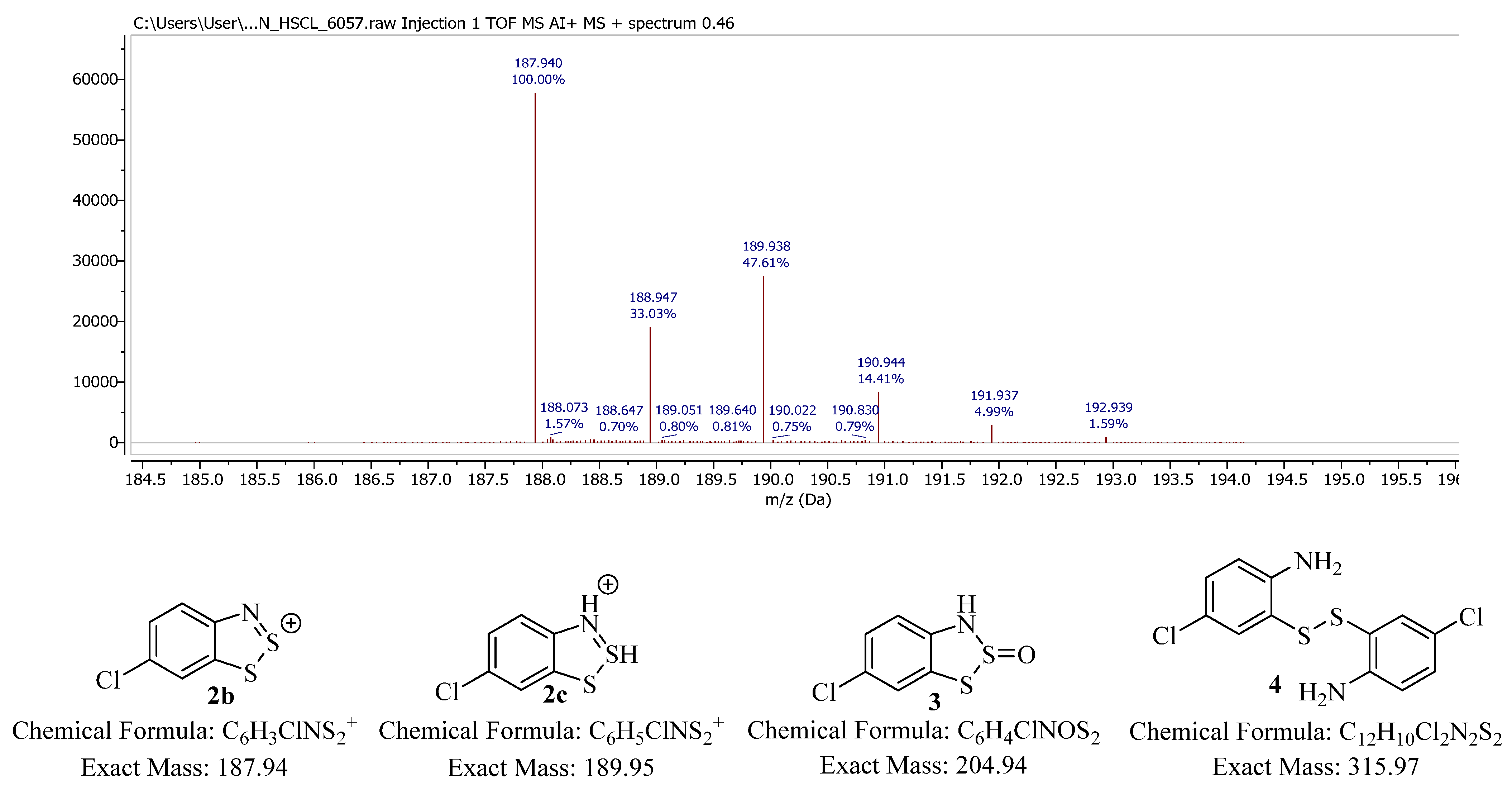
A closer inspection of the mass spectrum of
2 corresponds perfectly to the organic cation (
2b), confirming that the S–Cl interaction must be predominantly ionic in character (
Figure 3).
The Herz salt (
2) slowly reacts with moisture to form (
3,
Scheme 2) and this is accelerated by suspending the salt in an aqueous sodium acetate solution (5%) [
9]. The data for the
S-oxide compound (
3) has been comprehensively presented by Neo et al. [
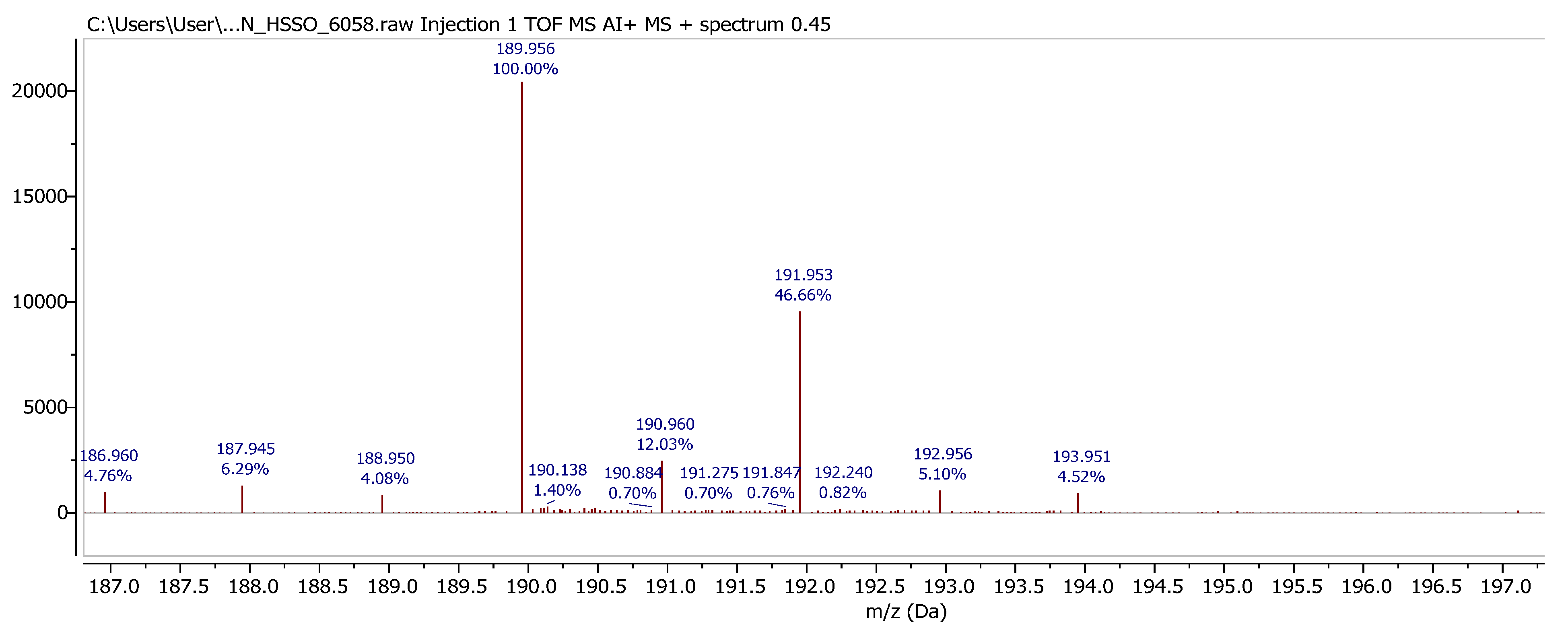
8] for a variety of derivatives, although it should be noted that conversion from 2 to 3 is readily monitored by ASAP-MS. Interestingly, the most intense ion observed from our solid sample of
3 is 189.95 mass units, not the expected mass of
3 but instead two units greater than the mass of
2, suggesting that a structure equating to
2c (
Figure 3) is actually the species the spectrometer detects (which could be flanked by an undetected counterion, e.g., a hydroxide).
The ASAP spectrum of compound
3 indicates that the sample contains impurities, which could not be removed effectively. One identified impurity is the ring-opened thiol
4, a product of additional hydrolysis and dimerization (
Scheme 2) [
7]. Indeed, when the derivatives of
3 have been reacted in the literature, it is generally as a synthetic equivalent of
4. Interestingly, for compound
3 the major observed ion (
Figure 4) does not correspond to the expected structure, but instead to a structure such as
2c (
Figure 3).
The following mechanism (
Scheme 3) was recently proposed and published, aiming to take into account the literature results as well as the data collected in this study [
6]. The consequences of the mechanism include the consumption of an excess of disulfur dichloride, although the precise extent of this is unclear. Regardless, when considering scale-up, this has severe consequences on the cost of the inputs and the amount of waste generated (not to mention the corrosive HCl fumes released from the reaction of S
2Cl
2). The Herz reaction is a remarkable transformation for its ability to not only introduce an
ortho-sulfur to an aniline ring, but also activate it to nucleophilic attack at the 4-position (via the introduced chloride); it is surprising that only a handful of publications have made use of this unique feature [
11,
12].
4. Materials and Methods
All solid-state Carbon-13 magic-angle spinning measurements were carried out at 100.6 MHz using a Bruker Avance III HD spectrometer (Karlsruhe, Germany) and 4 mm (rotor o.d.) probe. The spectra were acquired at a spin rate of 10 kHz. The cross-polarisation (CP) spectra were recorded with TOSS spinning sideband suppression, 1 ms contact time and with a recycle delay of 1 s. The recycle delay and contact time were optimised for each sample. Carbon spectral referencing is relative to neat tetramethylsilane and carried out by setting the high-frequency signal from an external sample of adamantane to 38.5 ppm.
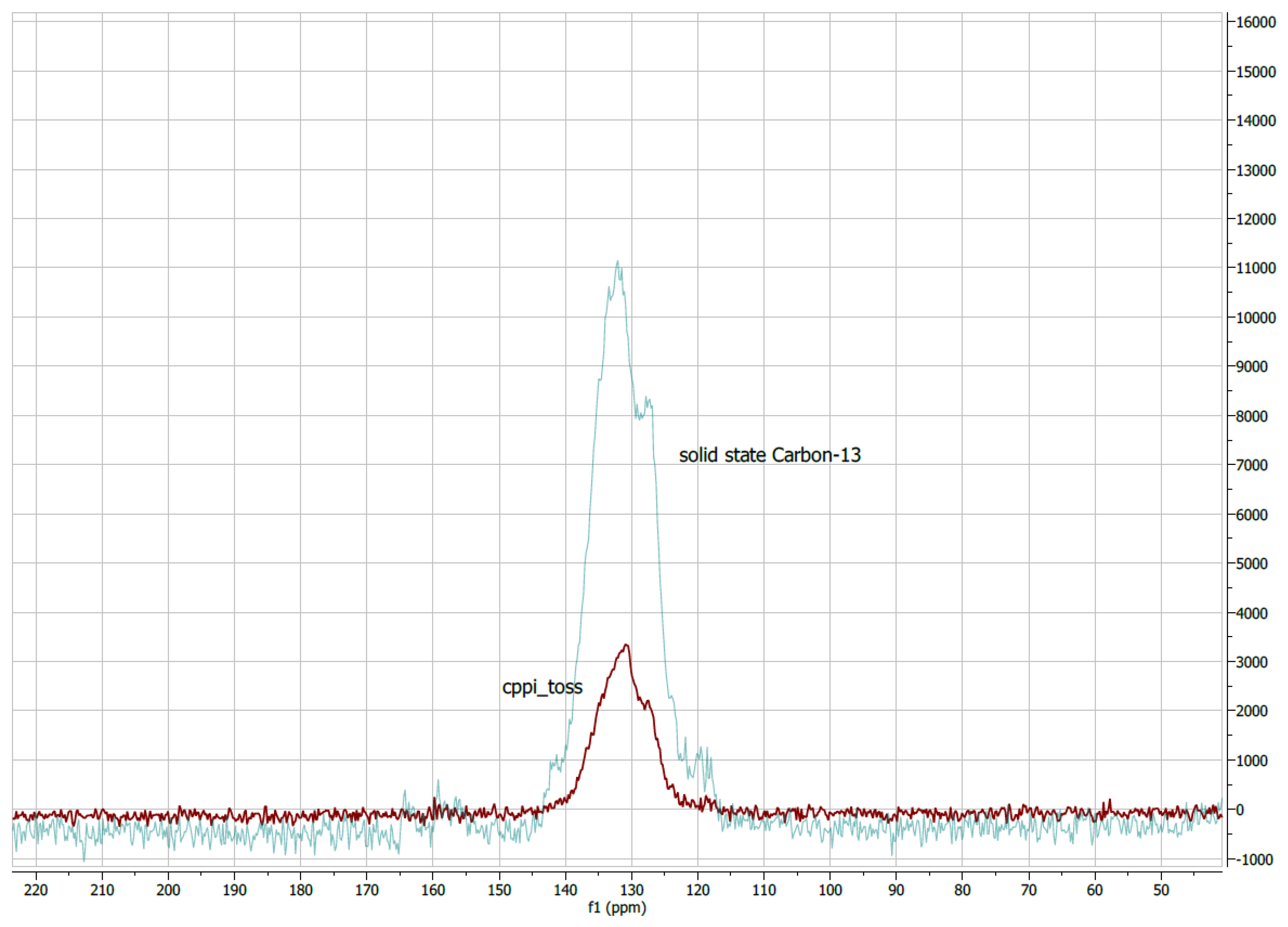
In addition to the CPTOSS spectrum, we used phase-inverted (cppi_toss) experiments to attempt to assist with identification and characterisation of the signals. This gave negative CH
2, null CH and positive C and CH
3 signals. Unfortunately, all of the carbon signals bunched tightly at approximately 130 ppm, meaning that assured identification was not possible (
Figure 5).
Aniline (1, 50 mmol, 4.65 g, 1 equiv.) was added to acetic acid (10 mL) in a flask surrounded by a water bath (ambient temperature, 21 °C). Disulfur dichloride (200 mmol, 32.4 g, 4 equiv.) was added dropwise, evacuating the emitted fumes. The mixture was stirred for 30 min at room temperature and then heated at 80 °C for 4 h. Toluene (125 mL) was added to precipitate the 6-chloro-3H-benzo[d][1,2,3]dithiazolium chloride (2) as a dark red powder, contaminated with elemental sulfur (10.8 g, 96% recovery and 61% purity based upon elemental analysis), isolated by filtration from a brown liquor and washed with dry toluene (50 mL) followed by dry dichloromethane (50 mL), then dried in vacuo. IR (neat) υmax/cm−1 3019 (m, N-H), 1705 (m, N-H), 1574 (s, C=N), 1499 (m, Ar C-C), 1302 (s), 1299 (s), 1120 (m), 1103 (m), 945 (m), 823 (m), 751 (s), 658 (m), 523.5 (w), 406.4 (m). ASAP-MS: Rt = 0.67 min, m/z 187.940 ([M]+, 100%). HR-MS: calculated for: 187.9395, C6H3NS235Cl, found: 187.9398 (Δ −1.6 ppm, −0.3 mDa). Elemental analysis found: C 20.06% (20.44), H 1.26% (0.86), N 3.38% (3.97).
Based upon the small-scale preparation, a large-scale synthesis was conducted.
A 2 L jacketed reactor connected to an external Huber Chiller set to maintain a working 15 °C internal temperature and fitted with a mechanical stirrer and nitrogen purge line (Purge outlet to NaOH scubber solution) was charged with acetic acid (200 mL). To the reactor was added aniline (1, 1 mol, 93.13 g, 1 equiv.), and the solution was stirred until an internal temperature of 15 °C was reached (~15 min). Disulfur dichloride (319.6 mL, 4 mol, 4 equiv.) was added over 1 h dropwise (pressure equalised dropping funnel). Liberation of gas accompanied by a strong exotherm, with temperature reaching and remaining at 25 °C for the duration of the controlled addition, was encountered. Following the addition, the mixture was stirred for an additional 45 min at 25 °C, and then the temperature was raised over 45 min and the mixture heated at 80 °C for 4 h. The mixture was cooled to 35 °C and diluted by the addition of a 8:2 mixture of toluene:DCM (1 L). The solution was further cooled to 10 °C, furnishing 6-chloro-3H-benzo[d][1,2,3]dithiazolium chloride (2) as a dark red suspension. The material was dispensed onto a sintered filter bed and agitated (nitrogen bubbling and stirring) with additional portions of toluene (2 × 500 mL) then dichloromethane (500 mL), with intermediate filtration. The solid was dried in vacuo at 40 °C for 8 h. The solid (213.9 g, 95% recovery and 91% purity based upon elemental analysis) had identical spectroscopic data to the small-scale reaction but indicated a higher overall purity and darker red colour. Elemental analysis found: C 29.21% (32.16), H 1.12% (1.35), N 5.88% (6.25).
6-chloro-3
H-benzo[
d][1,2,3]dithiazolium chloride (
2) (with the equivalent of 4S contamination as determined by elemental analysis) (31 mmol, 10.8 g, 1 equiv.) was suspended in a solution of sodium acetate trihydrate (62 mmol, 10.3 g, 2 equiv.) in water (125 mL) and stirred vigorously for 14 h. A purple solid was obtained by filtration from a mauve liquor and washed with water (50 mL) followed by EtOAc (50 mL). The purple solid was dissolved in boiling MeOH (800 mL), failing to crystallise and instead precipitating upon cooling to ambient temperature. A 6-chloro-3
H-benzo[
d][1,2,3]dithiazole-2-oxide contamination of S
8 (9.3 g, 94%) was obtained as a dark purple solid. Sulfur removal was attempted by washing the purple solid with hot THF using a Soxhlet procedure, but this had little impact on the sulfur content. The NMR data in CDCl
3 is identical to that reported previously [
8]. Greater solubility in DMSO allows a better quality of spectra:
1H NMR (400 MHz; DMSO-
d6) 7.50–7.68 (2H, m, Ar-H), 7.13–7.28 (1H, m, Ar-H), 7.18 (1H, s, N-H) [
8]. Solubility was insufficient for the resolved
13C NMR. IR (neat) υ
max/cm
−1 2785 (m, N-H), 2547 (m, N-H), 1600 (s, C=N), 1572 (m, Ar CC), 1467 (s), 1377 (s), 1098 (m), 1018 (m), 817 (m), 615 (m), 492 (s), 447 m. ASAP-MS: Rt = 0.44 min,
m/
z 189.955 ([M]
+, 100%). Accurate Mass found 189.9539. C
6H
5NS
2 35Cl = 189.9552. (−1.3 mDa, Δ 6.8 ppm). Melting point 178.1–182.4 °C (MeOH). Literature: 113–114 °C (in absence of sulfur, MeOH) [
8]. Elemental analysis found: C 21.55% (21.57), H 1.57% (1.21), N 3.56% (4.19). Solid state NMR data to be found in the
Supplementary Materials.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}