
N,N-bis(2-quinolinylmethyl)benzylamine


Abstract
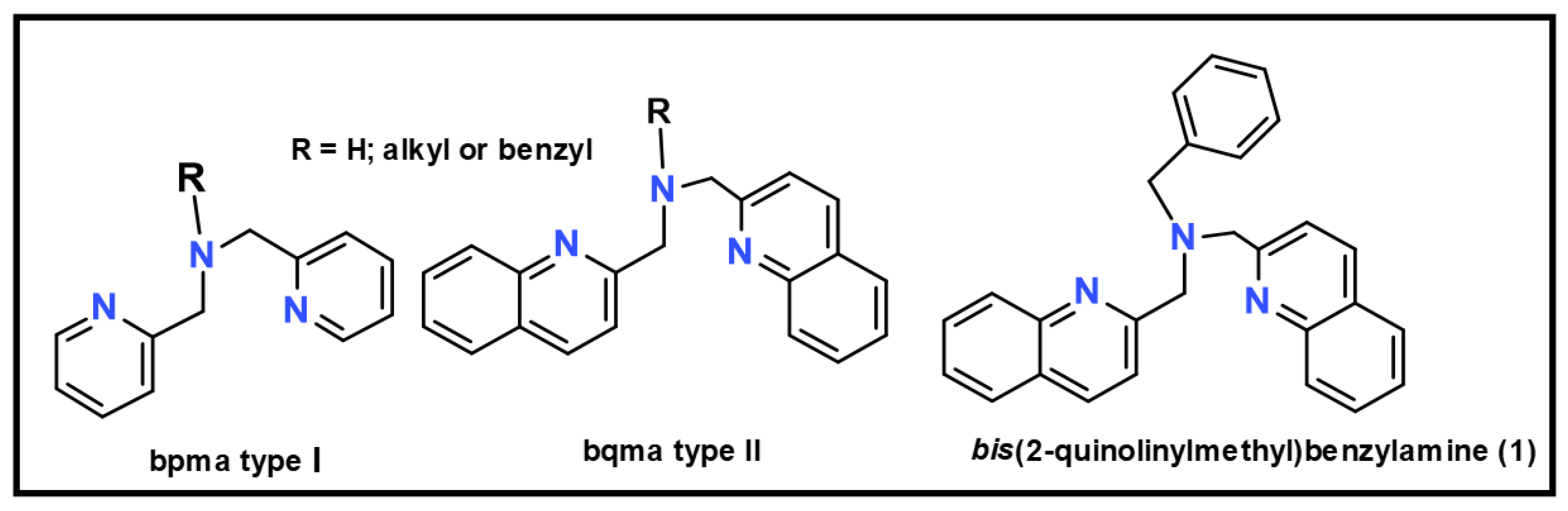
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of bis(2-quinolinylmethyl)benzylamine (1)
3.2. Spectroscopic Characterization of 1
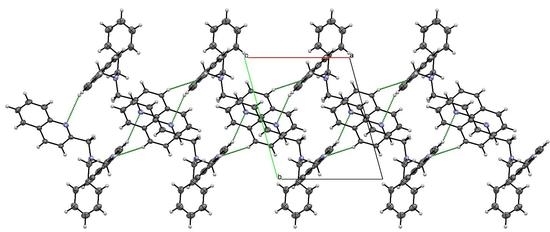
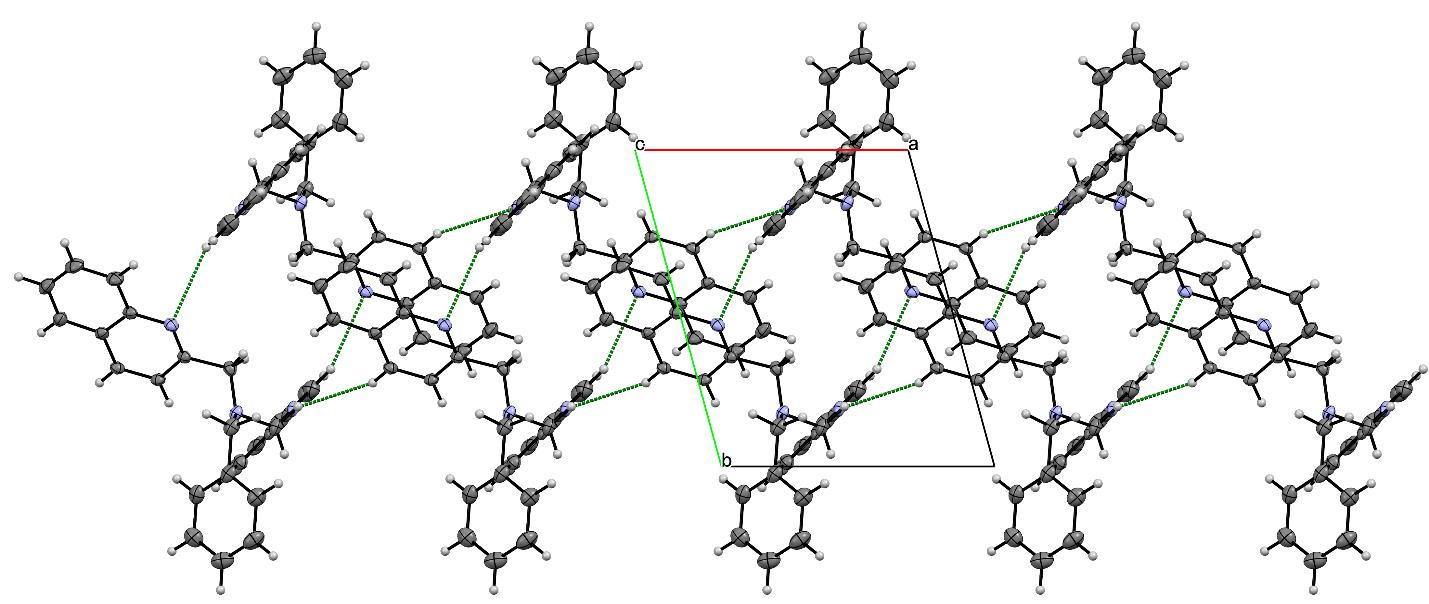
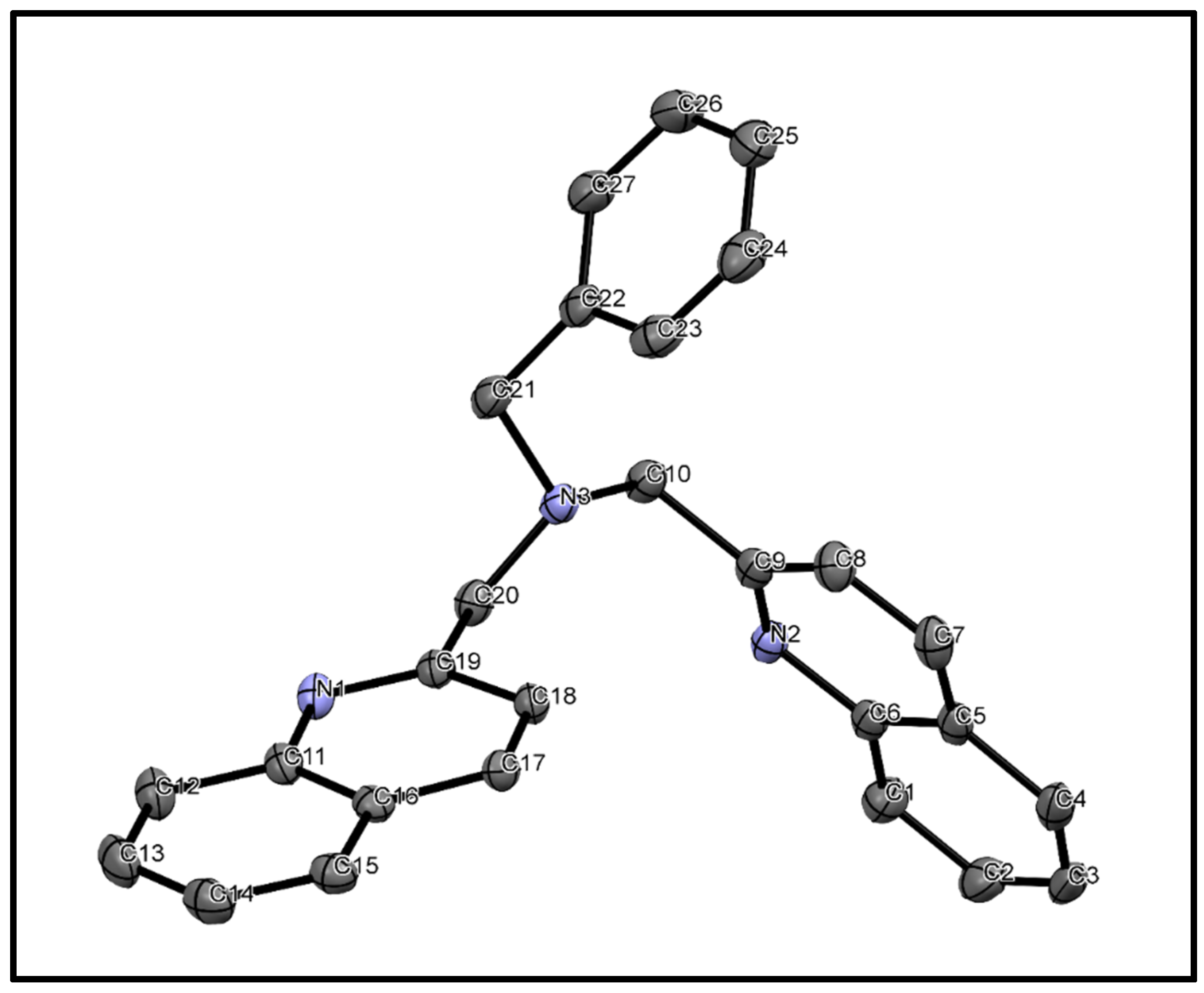
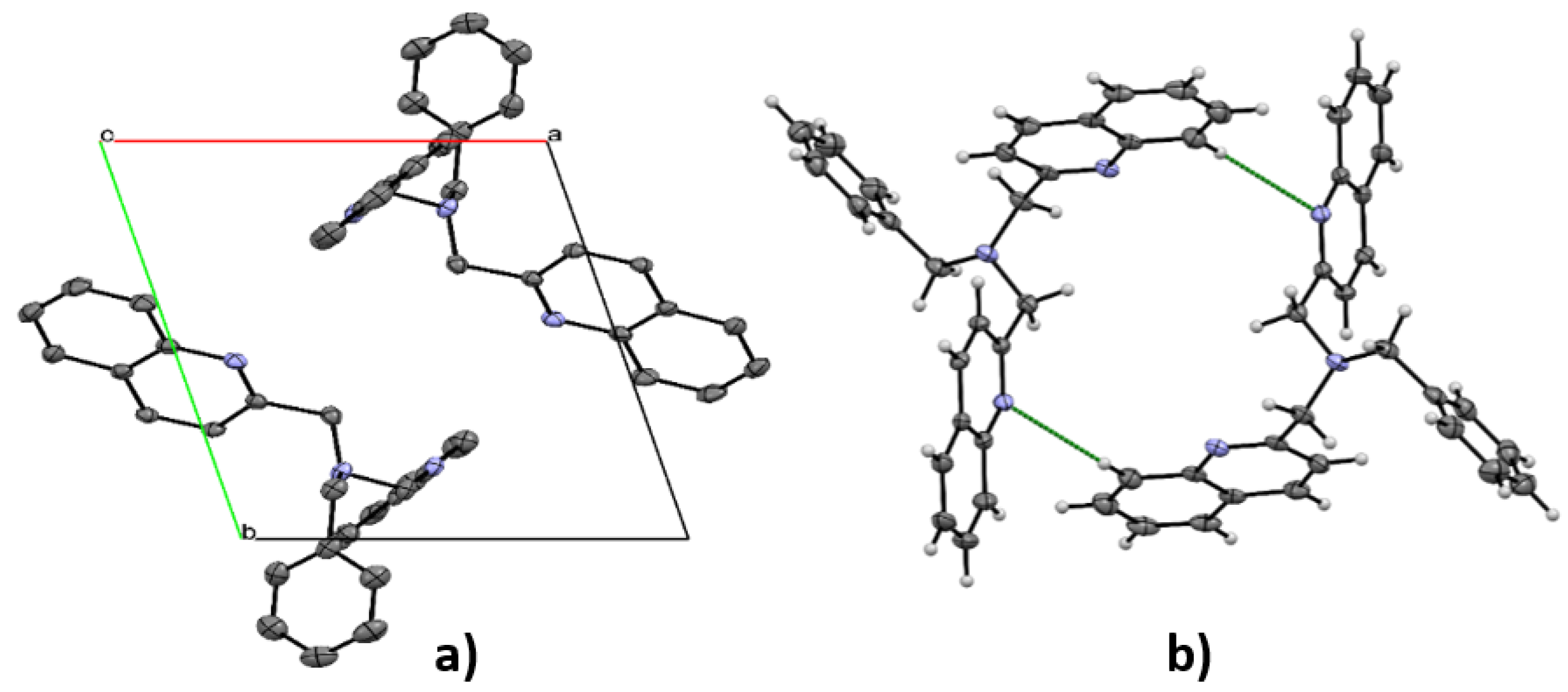
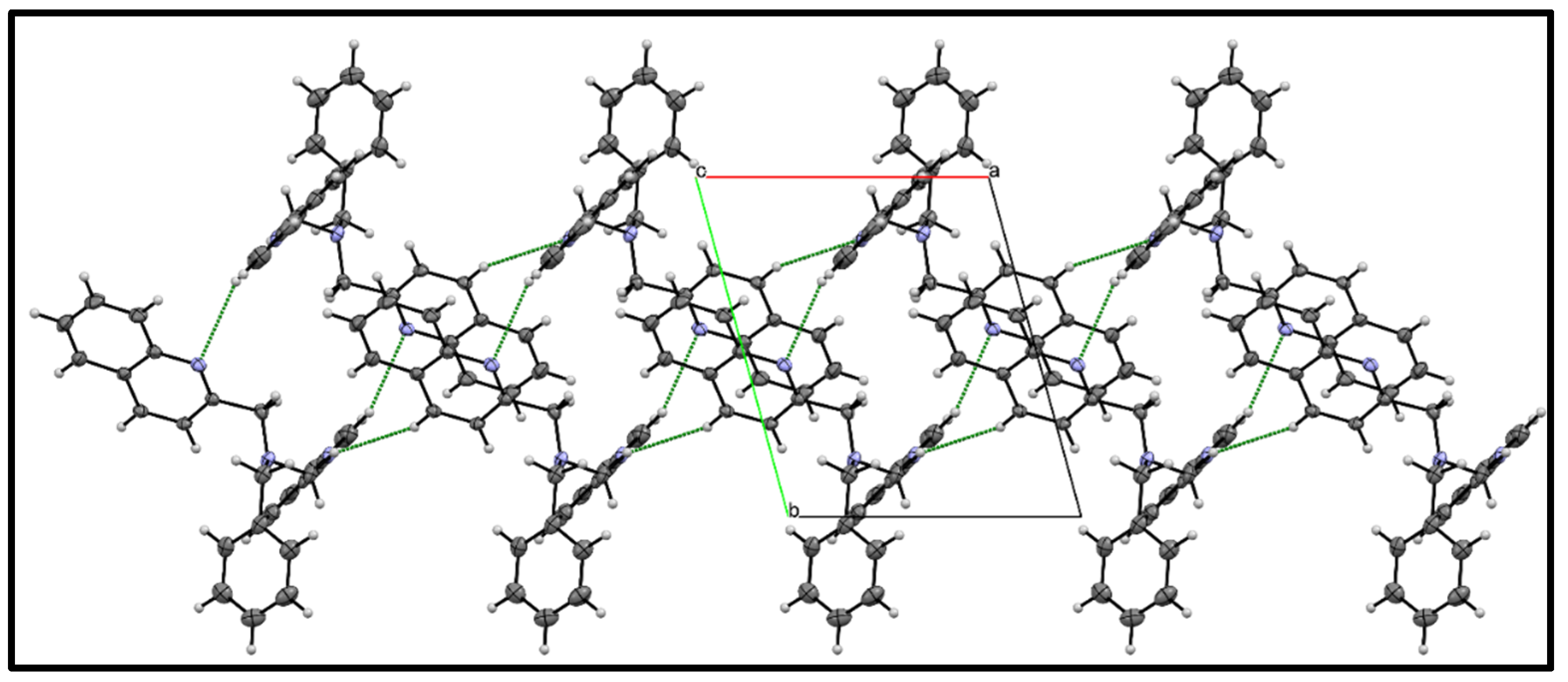
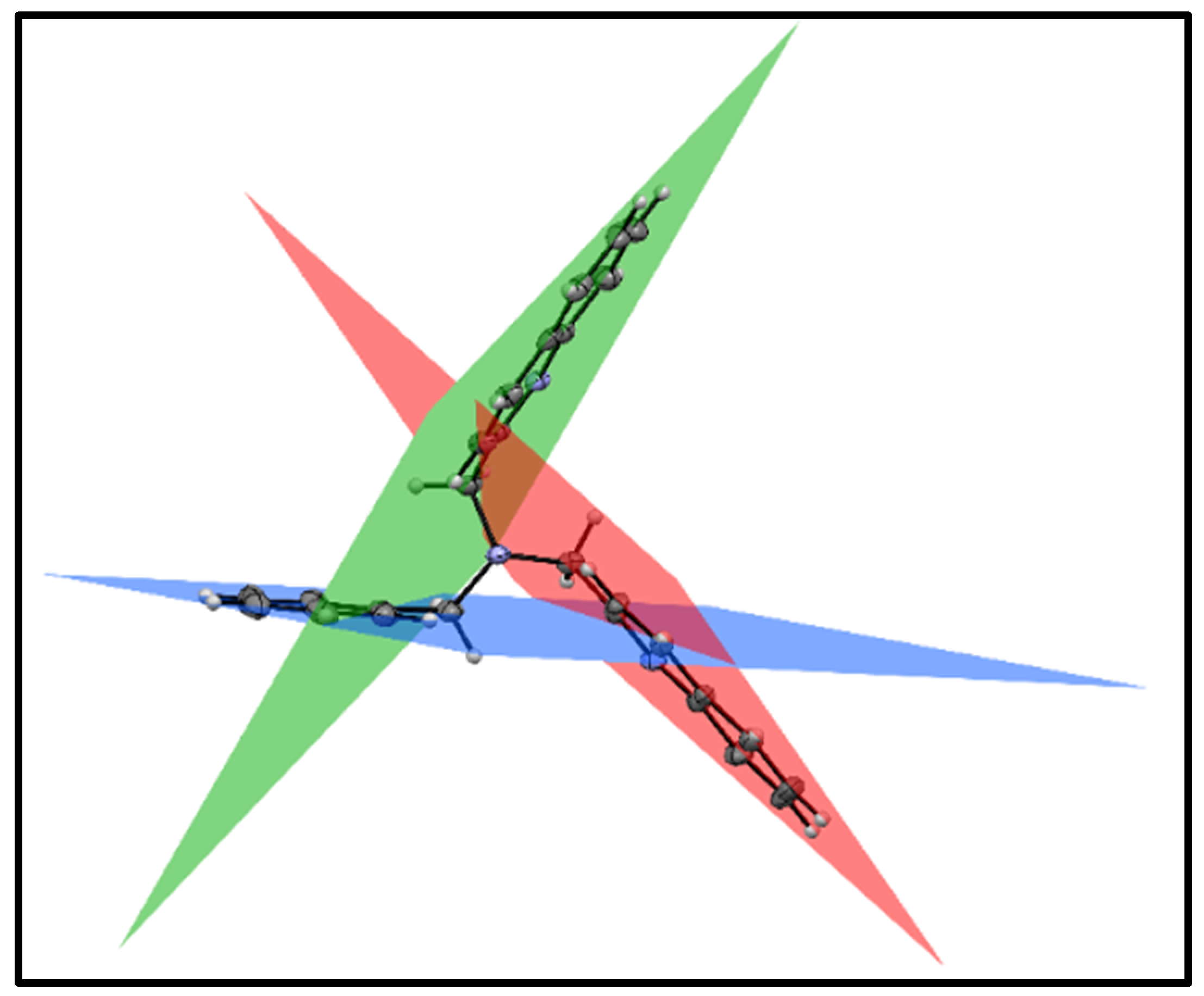
3.3. X-ray Diffraction
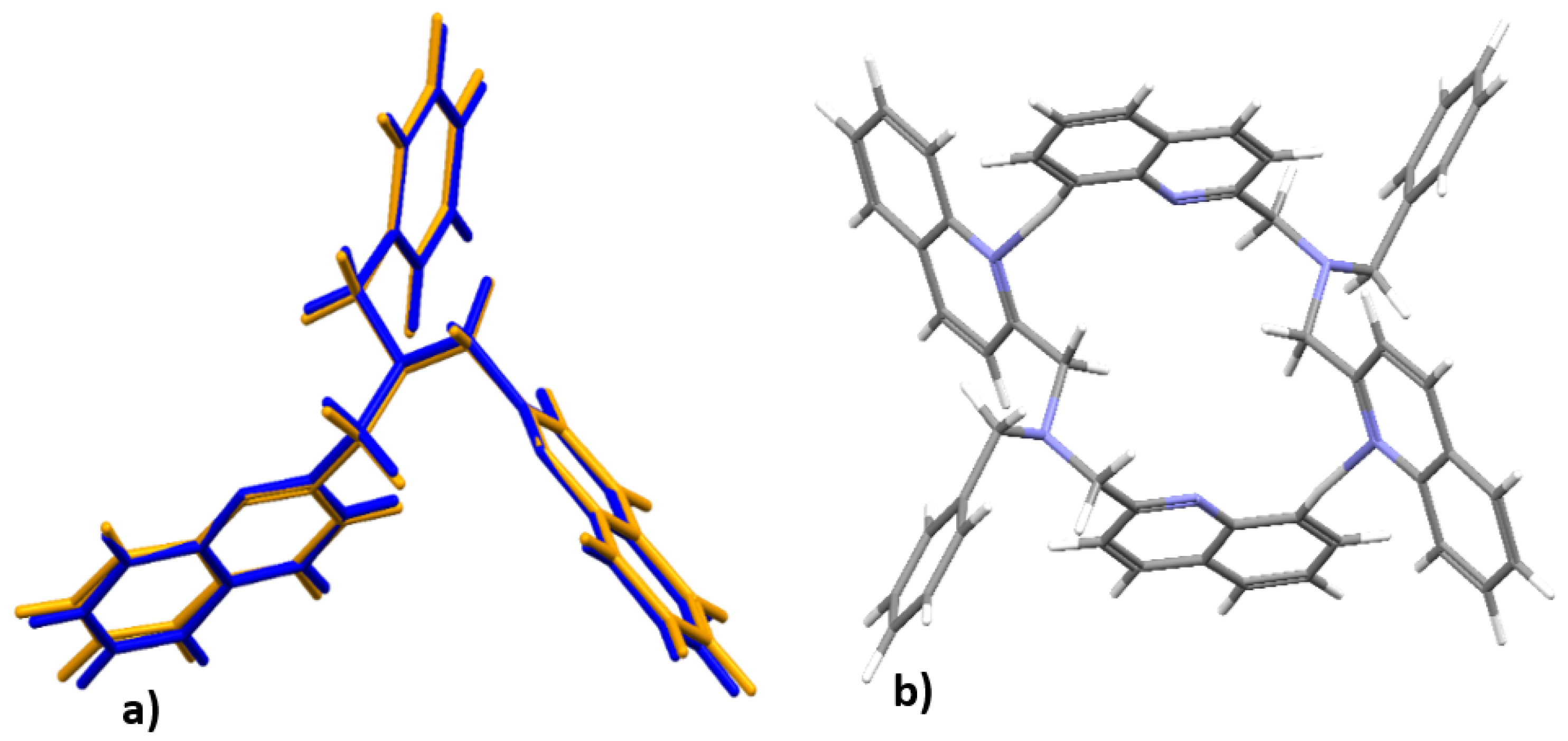
3.4. DFT Optimized Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| bqma | bis(2-quinolinylmethyl)benzylamine |
| H-bonds | hydrogen bonds |
| VdW | van der Waal |
References
- Kryatov, S.V.; Taktak, S.; Korendovych, I.V.; Rybak-Akimova, E.V. Dioxygen binding to complexes with FeII2(μ-OH)2 cores: Steric control of activation barrier and O2-adduct formation. Inorg. Chem. 2005, 44, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Kunishita, K.; Osako, T.; Tachi, Y.; Teraoka, J.; Itoh, S. Structural characterization of copper(I) complexes Supported by β-diketiminate ligands with different substitution patterns. Bull. Chem. Soc. Jpn. 2006, 79, 1729–1741. [Google Scholar] [CrossRef]
- Wei, N.; Murthy, N.N.; Chen, Q.; Zubieta, J.; Karlin, K.D. Copper(I)/Dioxygen Reactivity of Mononuclear Complexes with Pyridyl and Quinolyl Tripodal Tetradentate Ligands: Reversible Formation of Cu:O2 = 1:1 and 2:1 adducts. J. Inorg. Chem. 1994, 33, 1953–1965. [Google Scholar] [CrossRef]
- Lonnon, D.G.; Craig, D.C.; Colbran, S.B. Rhodium, palladium and platinum complexes of tris(pyridyla-lkyl)amine and tris(benzimidazolylmethyl)amine N-4-tripodal ligands. Dalton Trans. 2006, 3785–3797. [Google Scholar] [CrossRef]
- Li, J.L.; Jiang, L.; Wang, B.W.; Tian, J.L.; Gu, W.; Liu, X.; Yan, S.P. Significant differences in the biological activity of mononclear Cu(II) and Ni(II) complexes with the polyquinolinyl ligand. New J. Chem. 2015, 39, 529–538. [Google Scholar] [CrossRef]
- Bulatov, E.; Haukka, M. Non-conventional synthesis and photophysical studies of platinum(II) complexeswith methylene bridged 2,2’-dipyridylamine derivatives. Dalton Trans. 2019, 48, 3369–3379. [Google Scholar] [CrossRef]
- Mikata, Y.; Wakamatsu, M.; Kawamura, A.; Yamanaka, N.; Yano, S.; Odani, A.; Morihiro, K.; Tamotsu, S. Methoxy-Substituted TQEN Family of Fluorescent Zinc Sensors. Inorg. Chem. 2006, 42, 9262–9268. [Google Scholar] [CrossRef] [PubMed]
- Mikata, Y.; Sato, Y.; Takeuchi, S.; Kuroda, Y.; Konnoc, H.; Iwatsuki, S. Quinoline-based fluorescent zinc sensors with enhanced fluorescence intensity, Zn/Cd selectivity and metal-binding affinity by conformational restriction. Dalton Trans. 2013, 42, 9688–9698. [Google Scholar] [CrossRef] [PubMed]
- Mikata, Y.; Kizub, A.; Konno, H. TQPHEN (N,N,N′,N′-tetrakis(2-quinolylmethyl)-1,2-phenylenediamine) derivatives as highly selective fluorescent probes for Cd2+. Dalton Trans. 2015, 44, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Mikata, Y.; Kawata, K.; Iwatsuki, S.; Konno, H. Zinc-Specific Fluorescent Response of tris(isoquinolylmet-hyl)amines (isoTQAs). Inorg. Chem. 2012, 51, 1859–1865. [Google Scholar] [CrossRef]
- Ackerman, P.M.; Chipangura, M.; Mambanda, A.; Jaganyi, D. N,N-Bis(pyridin-2-ylmethyl)cyclohexanamine. Acta Cryst. 2012, E68, o2194–o2195. [Google Scholar] [CrossRef]
- Mambanda, A.; Jaganyi, D. A kinetics and mechanistic study on the role of the structural rigidity of the linker on the substitution reactions of chelated dinuclear Pt(II) complexes. Dalton Trans. 2012, 41, 908–920. [Google Scholar] [CrossRef]
- Akerman, M.P.; Chiazzari, V.A. An X-ray crystallographic and DFT study of the complementary hydrogen bonding of bidentate pyrrolide-imine Schiff base ligands. J. Mol. Struct. 2014, 1058, 22–30. [Google Scholar] [CrossRef]
- Barry, K.-L.; Grimmer, C.D.; Munro, O.Q.; Akerman, M.P. Self-assembled supramolecular structures of O,N,N′ tridentate imidazole–phenol Schiff base compounds. RSC Adv. 2020, 10, 7867–7878. [Google Scholar] [CrossRef]
- Kohn, W.A.D.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Andersson, M.P.; Uvdal, P. New Scale Factors for Harmonic Vibrational Frequencies Using the B3LYP Density Functional Method with the Triple-ζ Basis Set 6-311+G(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision E. 01; Gaussian, Inc.: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Oxford Diffraction, CrysAlis CCD and CrysAlis RED; Oxford Diffraction Ltd.: Abingdon, UK, 2008.
- 20. In Bruker, APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.D.; Wood, P.A. Mercury CSD 2.0. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code for 1 | l1_sq |
|---|---|
| Empirical formula | C27H23N3 |
| Formula weight | 389.48 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Triclinic |
| Space group | P-1 |
| Unit cell dimensions | a = 8.7296(6) Å, α = 70.914(2)° |
| b = 11.1447(6) Å, β = 86.955(2)° | |
| c = 12.3411(6) Å, γ = 74.653(2)° | |
| Volume | 1093.45(11) Å3 |
| Z | 2 |
| Density (calculated) | 1.183 Mg/m3 |
| Absorption coefficient | 0.070 mm−1 |
| F(000) | 412 |
| D | H | A | d(D—H), Å | d(H—A), Å | d(D—A), Å | ∠D—H…A,° |
|---|---|---|---|---|---|---|
| C′17 | H′17 | N2 | 0.950 | 2.732 | 3.594(2) | 151.30 |
| C′1 | H′1 | N1 | 0.950 | 2.749 | 3.584(2) | 147.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunter, L.A.; Naidoo, S.; Mambanda, A. N,N-bis(2-quinolinylmethyl)benzylamine. Molbank 2021, 2021, M1208. https://doi.org/10.3390/M1208
Hunter LA, Naidoo S, Mambanda A. N,N-bis(2-quinolinylmethyl)benzylamine. Molbank. 2021; 2021(2):M1208. https://doi.org/10.3390/M1208
Chicago/Turabian StyleHunter, Leigh A., Shivani Naidoo, and Allen Mambanda. 2021. "N,N-bis(2-quinolinylmethyl)benzylamine" Molbank 2021, no. 2: M1208. https://doi.org/10.3390/M1208
APA StyleHunter, L. A., Naidoo, S., & Mambanda, A. (2021). N,N-bis(2-quinolinylmethyl)benzylamine. Molbank, 2021(2), M1208. https://doi.org/10.3390/M1208