2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity


Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
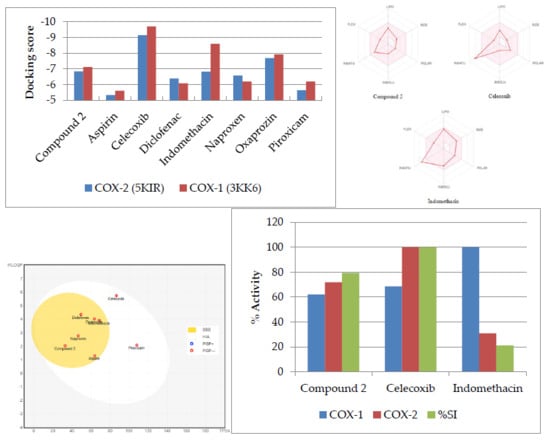
2.2. Molecular Docking
2.3. Physicochemical Properties and Drug-Likeness Analysis
2.4. In Vitro COX-1 and COX-2 Inhibitory Activity
3. Materials and Methods
3.1. General
3.2. Synthesis of 2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one (2)
3.3. Molecular Docking Studies
3.4. Determination of the Physicochemical Properties and Drug-Likeness
3.5. In Vitro COX-1 and COX-2 Inhibitory Activity Evaluation
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Asif, M. Diverse biologically active pyridazine analogs: A scaffold for the highly functionalized heterocyclic compounds. Rev. J. Chem. 2018, 8, 280–300. [Google Scholar] [CrossRef]
- Dubey, S.; Bhosle, P.A. Pyridazinone: An important element of pharmacophore possessing broad spectrum of activity. Med. Chem. Res. 2015, 24, 3579–3598. [Google Scholar] [CrossRef]
- Saini, M.; Mehta, D.K.; Das, R.; Saini, G. Recent advances in anti-inflammatory potential of pyridazinone derivatives. Mini Rev. Med. Chem. 2016, 16, 996–1012. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Sharma, D.; Bansal, R. Pyridazinone: An attractive lead for anti-inflammatory and analgesic drug discovery. Future Med. Chem. 2017, 9, 95–127. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Yang, G.; Xu, F.; Niu, Y.; Sun, Q.; Xu, P. Copper-catalyzed aerobic dehydrogenation of C–C to C=C bonds in the synthesis of pyridazinones. Eur. J. Org. Chem. 2013, 2013, 6130–6136. [Google Scholar] [CrossRef]
- Sircar, I. Substituted 6-Phenyl-3(2H)-pyridazinones Useful as Cardiotonic Agents. U.S. Patent 4404203, 13 September 1983. [Google Scholar]
- Islam, M.A.; Pillay, T.S. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2020, 38, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A. Pharmacoinformatics and molecular dynamic simulation studies to identify potential small-molecule inhibitors of WNK-SPAK/OSR1 signaling that mimic the RFQV motifs of WNK kinases. Arab. J. Chem. 2020, 13, 5107–5117. [Google Scholar] [CrossRef]
- Khan, A.; Diwan, A.; Thabet, H.K.; Imran, M.; Bakht, M.A. Discovery of novel pyridazine-based cyclooxygenase-2 inhibitors with a promising gastric safety profile. Molecules 2020, 25, 2002. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72 Pt 10, 772–776. [Google Scholar] [CrossRef]
- Amin, N.H.; Mohammed, A.A.; Abdellatif, K.R. Novel 4-methylsulfonylphenyl derivatives as NSAIDS with preferential COX-2 inhibition. Future Med. Chem. 2018, 10, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. Application of the SwissDrugDesign online resources in virtual screening. Int. J. Mol. Sci. 2019, 20, 4612. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.; Sharma, P.K.; Purohit, P. A journey of celecoxib from pain to cancer. Prostaglandins Other Lipid Mediat. 2020, 147, 106379. [Google Scholar] [CrossRef] [PubMed]
- Rimon, G.; Sidhu, R.S.; Lauver, D.A.; Lee, J.Y.; Sharma, N.P.; Yuan, C.; Frieler, R.A.; Trievel, R.C.; Lucchesi, B.R.; Smith, W.L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. USA 2010, 107, 28–33. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chain B of 5KIR (COX-2) | Chain B of 3KK6 (COX-1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| DS | RMSD | Ki | LE | LELP | LEScale | FQ | DS | RMSD | |
| 2 | −6.83 | 1.26 | 0.0067 | 0.40 | 6.62 | 0.47 | 0.85 | −7.11 | 0.99 |
| Aspirin | −5.33 | 1.49 | 0.020 | 0.41 | 3.12 | 0.53 | 0.77 | −5.60 | 1.28 |
| Celecoxib | −9.14 | 0.97 | 0.0012 | 0.35 | 9.71 | 0.36 | 0.97 | −9.69 | 1.23 |
| Diclofenac | −6.38 | 1.14 | 0.0093 | 0.34 | 10.76 | 0.44 | 0.77 | −6.07 | 1.29 |
| Indomethacin | −6.81 | 1.03 | 0.0068 | 0.27 | 13.44 | 0.37 | 0.73 | −8.59 | 1.04 |
| Naproxen | −6.57 | 1.50 | 0.0082 | 0.39 | 6.92 | 0.47 | 0.83 | −6.19 | 0.65 |
| Oxaprozin | −7.68 | 1.14 | 0.0036 | 0.35 | 9.71 | 0.40 | 0.87 | −7.91 | 0.61 |
| Piroxicam | −5.63 | 0.99 | 0.016 | 0.24 | 5.75 | 0.39 | 0.61 | −6.19 | 0.85 |
| Parameter | Compound 2 | Aspirin | Celecoxib | Diclofenac | Indomethacin | Naproxen | Oxaprozin | Piroxicam |
|---|---|---|---|---|---|---|---|---|
| NHA | 17 | 13 | 26 | 19 | 25 | 17 | 22 | 23 |
| MR | 76.96 | 44.90 | 89.96 | 77.55 | 96.12 | 66.95 | 83.73 | 87.52 |
| TPSA (Å) | 32.67 | 63.60 | 86.36 | 49.33 | 68.53 | 46.53 | 63.33 | 107.98 |
| Log P (o/w) (Consensus) | 2.65 | 1.19 ## | 3.40 | 4.51 ## | 4.27 ## | 3.18 ## | 4.19 ## | 3.06 ## |
| DL (BS) | Yes (0.55) | Yes (0.56) | Yes (0.55) | Yes (0.56) | Yes (0.56) | Yes (0.56) | Yes (0.56) | Yes (0.56) |
| WS | Moderate | Soluble | Moderate | Moderate | Moderate | Moderate | Moderate | Moderate |
| GI absorption | High (97.72%) # | High (87.05%) # | High (79.02%) # | High (91.98) # | High (85.35) # | High (92.94%) # | High (87.15%) # | High (71.75%) # |
| BBB permeant | Yes | Yes | No | Yes | Yes | Yes | Yes | No |
| P-gp substrate | No | No | No | No | No | No | No | No |
| CYP1A2 inhibitor | Yes | No | Yes | Yes | Yes | Yes | Yes | No |
| CYP2C19 inhibitor | Yes | No | No | Yes | Yes | No | Yes | No |
| CYP2C9 inhibitor | No | No | Yes | Yes | Yes | No | Yes | Yes |
| CYP2D6 inhibitor | No | No | No | Yes | No | No | Yes | No |
| CYP3A4 inhibitor | No | No | No | No | No | No | No | No |
| SP (cm/s) | −6.03 | −6.55 | −6.21 | −4.98 | −5.45 | −5.60 | −5.11 | −6.15 |
| Compound | COX-1 (IC50 in nM) | % COX-1 inhibition | COX-2 (IC50 in nM) | % COX-2 inhibition | SI | %SI |
|---|---|---|---|---|---|---|
| 2 | 368 ± 0.22 * | 61.95% | 25.13 ± 0.31 * | 71.78% | 14.64 | 79.34% |
| Celecoxib | 333 ± 0.12 * | 68.46% | 18.04 ± 0.22 * | 100.0% | 18.45 | 100.0% |
| Indomethacin | 228 ± 0.16 * | 100.0% | 58.41 ± 0.18 * | 30.88% | 3.90 | 21.13% |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, M. 2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity. Molbank 2020, 2020, M1155. https://doi.org/10.3390/M1155
Imran M. 2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity. Molbank. 2020; 2020(3):M1155. https://doi.org/10.3390/M1155
Chicago/Turabian StyleImran, Mohd. 2020. "2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity" Molbank 2020, no. 3: M1155. https://doi.org/10.3390/M1155
APA StyleImran, M. (2020). 2-Butyl-6-phenyl-4,5-dihydropyridazin-3(2H)-one: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity. Molbank, 2020(3), M1155. https://doi.org/10.3390/M1155