Synthesis and Spectral Characterization of 4,7-Dichloro-6-nitroquinazoline



Abstract
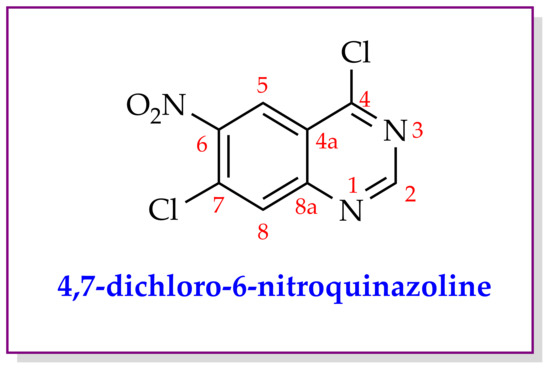
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedure
3.2.1. Preparation of 7-Chloroquinazolin-4(3H)-one (II)
3.2.2. Preparation of 7-Chloro-6-nitroquinazolin-4(3H)-one (7-Chloro-4-hydroxy-6-nitroquinazoline, III)
3.2.3. Preparation of 4,7-Dichloro-6-nitroquinazoline (IV)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia; Ishtiaq, M.; Khan, M. Quinazoline and quinazolinone as important medicinal scaffolds: A comparative patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Gatadi, S.; Lakshmi, T.V.; Nanduri, S. 4(3H)-Quinazolinone derivatives: Promising antibacterial drug leads. Eur. J. Med. Chem. 2019, 170, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Shagufta; Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm 2017, 8, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Buonerba, C.; Iaccarino, S.; Dolce, P.; Pagliuca, M.; Izzo, M.; Scafuri, L.; Costabile, F.; Riccio, V.; Ribera, D.; Mucci, B.; et al. Predictors of outcomes in patients with EGFR-mutated non-small cell lung cancer receiving EGFR tyrosine kinase inhibitors: A systematic review and meta-analysis. Cancers 2019, 11, 1259. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Keating, G.M. Afatinib in advanced NSCLC: A profile of its use. Drugs Ther. Perspect. 2018, 34, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Boehringer Ingelheim International GmbH. Available online: https://www.boehringer-ingelheim.com/oncology/lung-cancer/giotrif-gilotrif-afatinib-approved-nsclc (accessed on 4 February 2020).
- Himmelsbach, F.; Langkopf, E.; Blech, S.; Jung, B.; Baum, A.; Solca, F. Quinazoline Derivatives, Medicaments Containing Said Compounds, Their Utilization and Method for the Production Thereof. PCT Patent WO200250043A1, 27 June 2002. [Google Scholar]
- Soyka, R.; Rall, W.; Schnaubelt, J.; Sieger, P.; Kulinna, C. Process for Preparing Amino Crotonyl Compounds. U.S. Patent 20050085495A1, 21 April 2005. [Google Scholar]
- Schroeder, J.; Dziewas, G.; Fachinger, T.; Jaeger, B.; Reichel, C.; Renner, S. Process for Preparing Aminocrotonylamino-Substituted Quinazoline Derivatives. U.S. Patent US8188274B2, 29 May 2012. [Google Scholar]
- Xu, X. Method for Preparing Afatinib and Intermediate Thereof. PCT Patent WO2014180271A1, 13 November 2014. [Google Scholar]
- Xu, X. Afatinib and Preparation Method of Intermediate Thereof. PCT Patent WO2014183560A1, 20 November 2014. [Google Scholar]
- Ding, H.X.; Leverett, C.A.; Kyne, R.E., Jr.; Liu, K.K.C.; Fink, S.J.; Flick, A.C.; O’Donnell, C.J. Synthetic approaches to the 2013 new drugs. Bioorg. Med. Chem. 2015, 23, 1895–1922. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.J. Quinazoline Derivatives and Their Use as Anti-Cancer Agents. European Patent EP0635498A1, 25 January 1995. [Google Scholar]
- Barker, A.J. Quinazoline Derivative. PCT Patent WO1996033981A1, 17 May 1996. [Google Scholar]
- Brown, D.S.; Morris, J.J.; Thomas, A.P. Aniline Derivatives. PCT Patent WO1996015118A1, 23 May 1996. [Google Scholar]
- Barker, A.J. Quinazoline Derivatives. PCT Patent WO9616960A1, 6 June 1996. [Google Scholar]
- Thomas, A.P.; Hennequin, L.F.A.; Johnstone, C.; Stokes, E.S.E.; Lohmann, J.-J.M.; Clayton, E. Quinazoline Derivatives and Pharmaceutical Compositions Containing Them. PCT Patent WO1998013354A1, 2 April 1998. [Google Scholar]
- Barker, A.J. Quinazoline Derivative. U.S. Patent US5952333A, 14 September 1999. [Google Scholar]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Matsumoto, M.; Obara, F.; Nagasaki, T.; Hayashi, H. Structure-activity relationships of quinazoline derivatives: Dual-acting compounds with inhibitory activities toward both TNF-α production and T cell proliferation. Bioorg. Med. Chem. Lett. 2001, 11, 545–548. [Google Scholar] [CrossRef]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukazawa, T.; Hayashi, H. Discovery of quinazolines as a novel structural class of potent inhibitors of NF-κB activation. Bioorg. Med. Chem. 2003, 11, 383–391. [Google Scholar] [CrossRef]
- Matsuno, K.; Seishi, T.; Nakajima, T.; Ichimura, M.; Giese, N.A.; Yu, J.-C.; Oda, S.; Nomoto, Y. Potent and selective inhibitors of platelet-derived growth factor receptor phosphorylation. Part 4: Structure-activity relationships for substituents on the quinazoline moiety of 4-[4-(N-substituted(thio)carbamoyl)-1-piperazinyl]-6,7-dimethoxyquinazoline derivatives. Bioorg. Med. Chem. Lett. 2003, 13, 3001–3004. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.K.; Lee, S.; Choi, N.S.; Lee, J.K.; Moon, S.K.; Choi, H.; Kim, S.J.; Kim, Y.H.; Kang, S.K.; Lee, H.W.; et al. Quinazoline Derivative as Phosphodiesterase Inhibitor and a Process for Preparing the Same. PCT Patent WO2008020711A1, 21 February 2008. [Google Scholar]
- Guo, J.; Wang, M.; Jiang, Y.; Zhang, X. Quinazoline Derivatives, Preparation Methods and Uses Thereof. European Patent EP1990337A1, 12 November 2008. [Google Scholar]
- Jiang, Y.; Guo, J. Medical Application of 4-Anilinoquinazoline Derivatives. CN Patent CN101347433A, 21 January 2009. [Google Scholar]
- Tang, T.; Yu, L.; Feng, H.; Yan, Q.; Wang, B.; Wang, Z.; Zhu, D.; Chen, H. Heterocyclic Compound of 4-Aminoquinazoline Useful in Treatment of Cancer and Its Application. CN Patent CN102942561A, 27 February 2013. [Google Scholar]
- Zhang, K.; Cao, D.; Xue, N.; Shi, Q.; Du, Y.; Dong, M. Phenylurea-Quinazoline Coupling Compounds: Preparation Method, Medicine Composition and Medicinal Use. CN Patent CN103382182A, 6 November 2013. [Google Scholar]
- Zhang, X.; Peng, T.; Ji, X.; Li, J.; Tong, L.; Li, Z.; Yang, W.; Xu, Y.; Li, M.; Ding, J.; et al. Design, synthesis and biological evaluation of novel 4-anilinoquinazolines with C-6 urea-linked side chains as inhibitors of the epidermal growth factor receptor. Bioorg. Med. Chem. 2013, 21, 7988–7998. [Google Scholar] [CrossRef] [PubMed]
- Buha, V.M.; Rana, D.N.; Chhabria, M.T.; Chikhalia, K.H.; Mahajan, B.M.; Brahmkshatriya, P.S.; Shah, N.K. Synthesis, biological evaluation and QSAR study of a series of substituted quinazolines as antimicrobial agents. Med. Chem. Res. 2013, 22, 4096–4109. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.; Li, X.; Zhang, X.; Liu, Y.; Gong, N.; Zhou, Y.; Chen, K. Artemisinin Derivatives: Preparation Process and Application. PCT Patent WO2014023081A1, 13 February 2014. [Google Scholar]
- Tang, T.; Yu, L.; Feng, H.; Yan, Q.; Wang, B.; Wang, Z.; Zhu, D.; Chen, H. 4-Quinazolinamine Heterocyclic Compound and Use Thereof. PCT Patent WO2014071824A1, 15 May 2014. [Google Scholar]
- Yuan, J.; Han, N.; Yi, H.; Wang, Y.; Yang, S.; Wong, J.C. Potent Small Molecule Inhibitors of Autophagy, and Methods of Use Thereof. PCT Patent WO2014145512A3, 31 December 2014. [Google Scholar]
- Lai, Y.; Pang, J.; Luo, M.; Chen, F.; Wang, P.; Zhang, Y. Preparation of α-Cyano-α,β-Unsaturated Amides as Antitumor Agents. CN Patent CN104774184A, 15 July 2015. [Google Scholar]
- Zhang, K.; Cao, D.; Xue, N.; Shi, X.; Lu, K.; Gu, J.; Wang, L. Aryl Formyl Urea-Coupled Quinazoline Compound Useful in Treatment of Cancer and Its Preparation. CN Patent CN105753793A, 13 July 2016. [Google Scholar]
- Min, J.; Guo, K.; Suryadevara, P.K.; Zhu, F.; Holbrook, G.; Chen, Y.; Feau, C.; Young, B.M.; Lemoff, A.; Connelly, M.C.; et al. Optimization of a novel series of ataxia-telangiectasia mutated kinase inhibitors as potential radiosensitizing agents. J. Med. Chem. 2016, 59, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Tu, Z.; Hao, H.; Yao, Y.; Qiu, Y.; Yao, H.; Qiang, L.; Chen, D. A Kind of Benzo-Aza Virtue Cyclics and Its Preparation Method and Application. CN Patent CN107674059A, 9 February 2018. [Google Scholar]
- Bridges, A.J.; Zhou, H.; Cody, D.R.; Rewcastle, G.W.; McMichael, A.; Showalter, H.D.H.; Fry, D.W.; Kraker, A.J.; Denny, W.A. Tyrosine kinase inhibitors. 8. An unusually steep structure−activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor. J. Med. Chem. 1996, 39, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.T.; Wang, G.F.; Yang, Y.Q.; Jin, F.; Wang, Y.; Xie, X.Y.; Mach, R.H.; Huang, Y.S. Synthesis and pharmacological evaluation of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives as sigma-2 receptor ligands. Eur. J. Med. Chem. 2018, 147, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H. The Chemistry of Imidoyl Halides; Plenum Press: New York, NY, USA, 1968; pp. 55–56. [Google Scholar]
- Li, H.G.; Kim, C.K.; Lee, B.S.; Kim, C.K.; Rhee, S.K.; Lee, I. Nucleophilic substitution at the imidoyl carbon atom: Intermediate mechanistic and reactivity behavior between carbonyl and vinyl carbon substitution. J. Am. Chem. Soc. 2001, 123, 2326–2333. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.J.; Bilodeau, M.T. A mild method for the formation and in situ reaction of imidoyl chlorides: Conversion of pyridine-1-oxides to 2-aminopyridine amides. Org. Lett. 2002, 4, 3127–3129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, X.; Wang, W.; Xiao, Y. Preparation Method for Afatinib Intermediate 6-Nitro-7-Chloro-4-Quinazolinone. CN Patent CN105712940A, 29 June 2016. [Google Scholar]
- Abraham, R.J.; Byrne, J.J.; Griffiths, L.; Perez, M. 1H chemical shifts in NMR: Part 23, the effect of dimethyl sulphoxide versus chloroform solvent on 1H chemical shifts. Magn. Reson. Chem. 2006, 44, 491–509. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
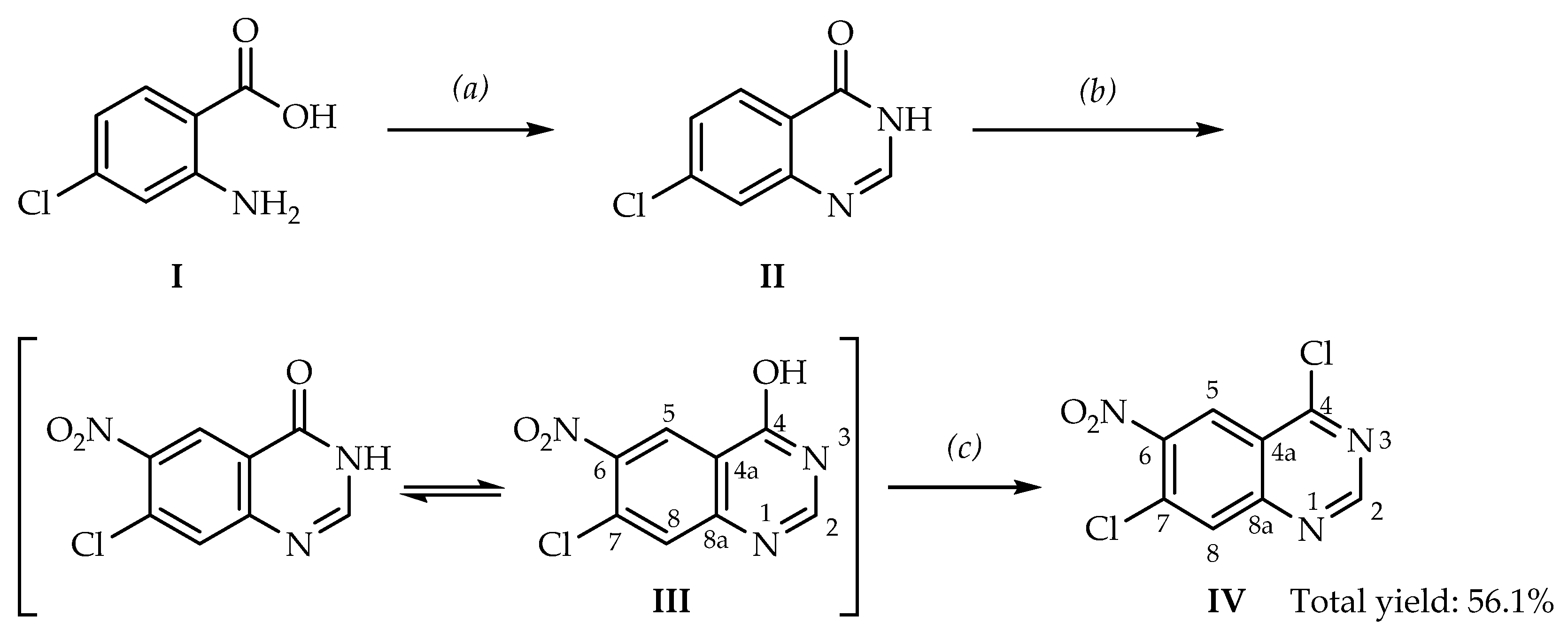{kind=link}
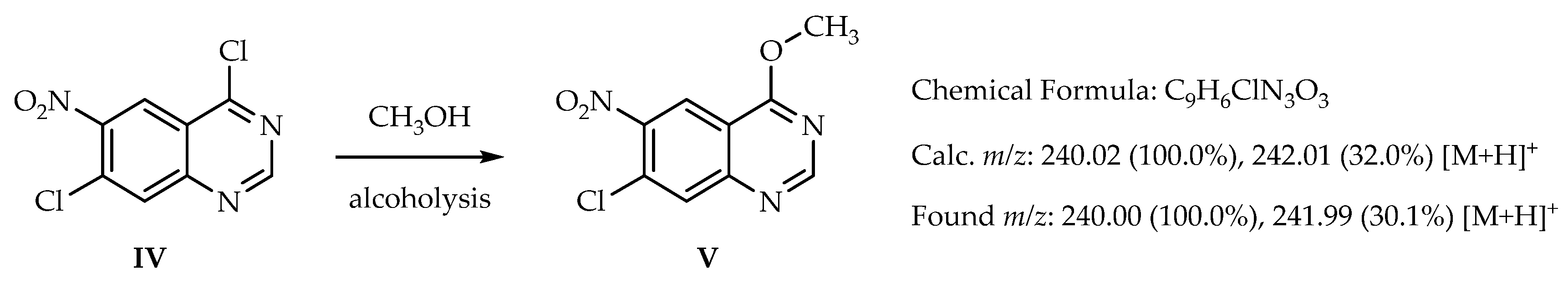{kind=link}
| Signals | δ (ppm) for Compound III | δ (ppm) for Compound IV | |||||
|---|---|---|---|---|---|---|---|
| Ref [30] (in DMSO-d6) | Ref [43] (in DMSO-d6) | Ref [39] (in DMSO-d6) | Our Data (in DMSO-d6) | Ref [38] (in DMSO-d6) | Ref [37] (in DMSO-d6) | Our Data (in CDCl3) | |
| OH or NH forms | 3.30 * (br.s, 1H, NH form) | 12.79 (br.s, 1H, OH form) | 12.79 (br.s, 1H, OH form) | 12.73 (br.s, 1H, OH form) | NA ** | NA | NA |
| H-5 | 8.53 (s, 1H) | 8.69 (s, 1H) | 8.67 (s, 1H) | 8.64 (s, 1H) | 8.60 (s, 1H) | 9.56 (s, 1H) | 9.18 (s, 1H) |
| H-2 | 8.28 (s, 1H) | 8.32 (s, 1H) | 8.31 (s, 1H) | 8.27 (s, 1H) | 8.27 (s, 1H) | 8.71 (s, 1H) | 8.76 (s, 1H) |
| H-8 | 7.97 (s, 1H) | 8.03 (s, 1H) | 8.01 (s, 1H) | 7.97 (s, 1H) | 7.85 (s, 1H) | 8.28 (s, 1H) | 8.30 (s, 1H) |
| Signals | δ (ppm) for Compound III | δ (ppm) for Compound IV | ||
|---|---|---|---|---|
| Literature | Our Data (in DMSO-d6) | Ref [38] (in DMSO-d6) | Our Data (in CDCl3) | |
| C-4 | NA | 159.3 | 159.0 | 163.6 |
| C-2 | NA | 151.5 | 150.3 | 156.9 |
| C-8a | NA | 149.6 | 149.2 | 151.6 |
| C-6 | NA | 144.7 | 144.8 | 147.5 |
| C-7 | NA | 130.4 | 131.1 | 132.8 |
| C-8 | NA | 129.4 | 129.4 | 132.2 |
| C-5 | NA | 124.2 | 124.2 | 123.5 |
| C-4a | NA | 121.7 | 121.4 | 122.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.N.; Tran, T.H.; Dao, N.S.H.; Nguyen, V.G.; Nguyen, D.L.; Trinh, N.T.; Nguyen, V.H. Synthesis and Spectral Characterization of 4,7-Dichloro-6-nitroquinazoline. Molbank 2020, 2020, M1134. https://doi.org/10.3390/M1134
Nguyen TN, Tran TH, Dao NSH, Nguyen VG, Nguyen DL, Trinh NT, Nguyen VH. Synthesis and Spectral Characterization of 4,7-Dichloro-6-nitroquinazoline. Molbank. 2020; 2020(2):M1134. https://doi.org/10.3390/M1134
Chicago/Turabian StyleNguyen, Thi Ngoc, Thi Huong Tran, Nguyet Suong Huyen Dao, Van Giang Nguyen, Dinh Luyen Nguyen, Nguyen Trieu Trinh, and Van Hai Nguyen. 2020. "Synthesis and Spectral Characterization of 4,7-Dichloro-6-nitroquinazoline" Molbank 2020, no. 2: M1134. https://doi.org/10.3390/M1134
APA StyleNguyen, T. N., Tran, T. H., Dao, N. S. H., Nguyen, V. G., Nguyen, D. L., Trinh, N. T., & Nguyen, V. H. (2020). Synthesis and Spectral Characterization of 4,7-Dichloro-6-nitroquinazoline. Molbank, 2020(2), M1134. https://doi.org/10.3390/M1134