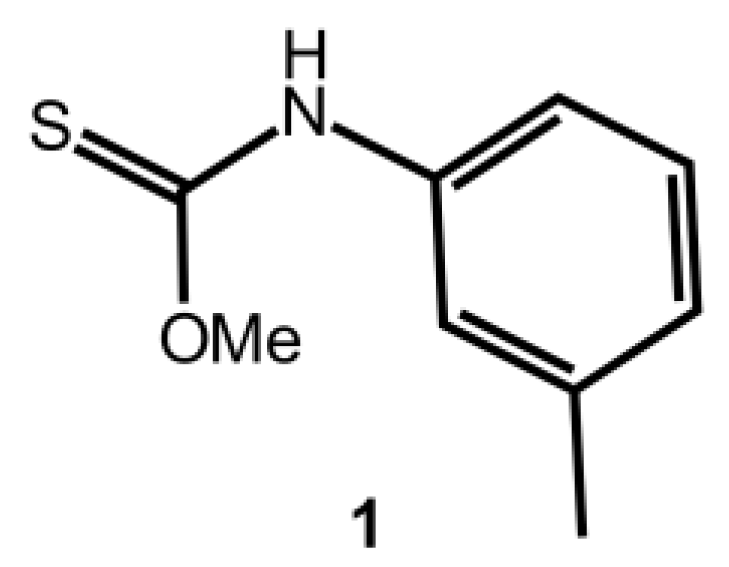
O-Methyl m-Tolylcarbamothioate



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
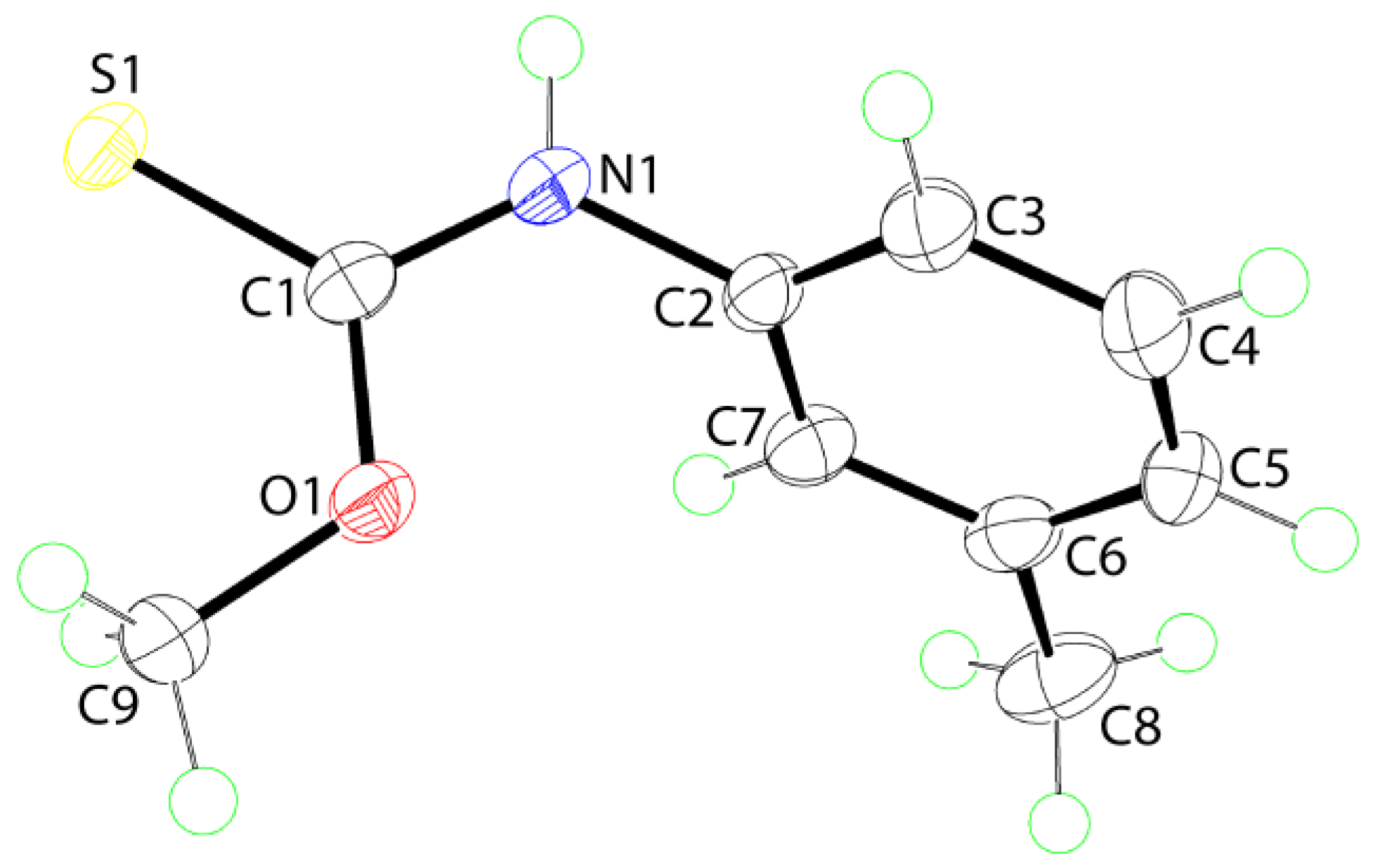
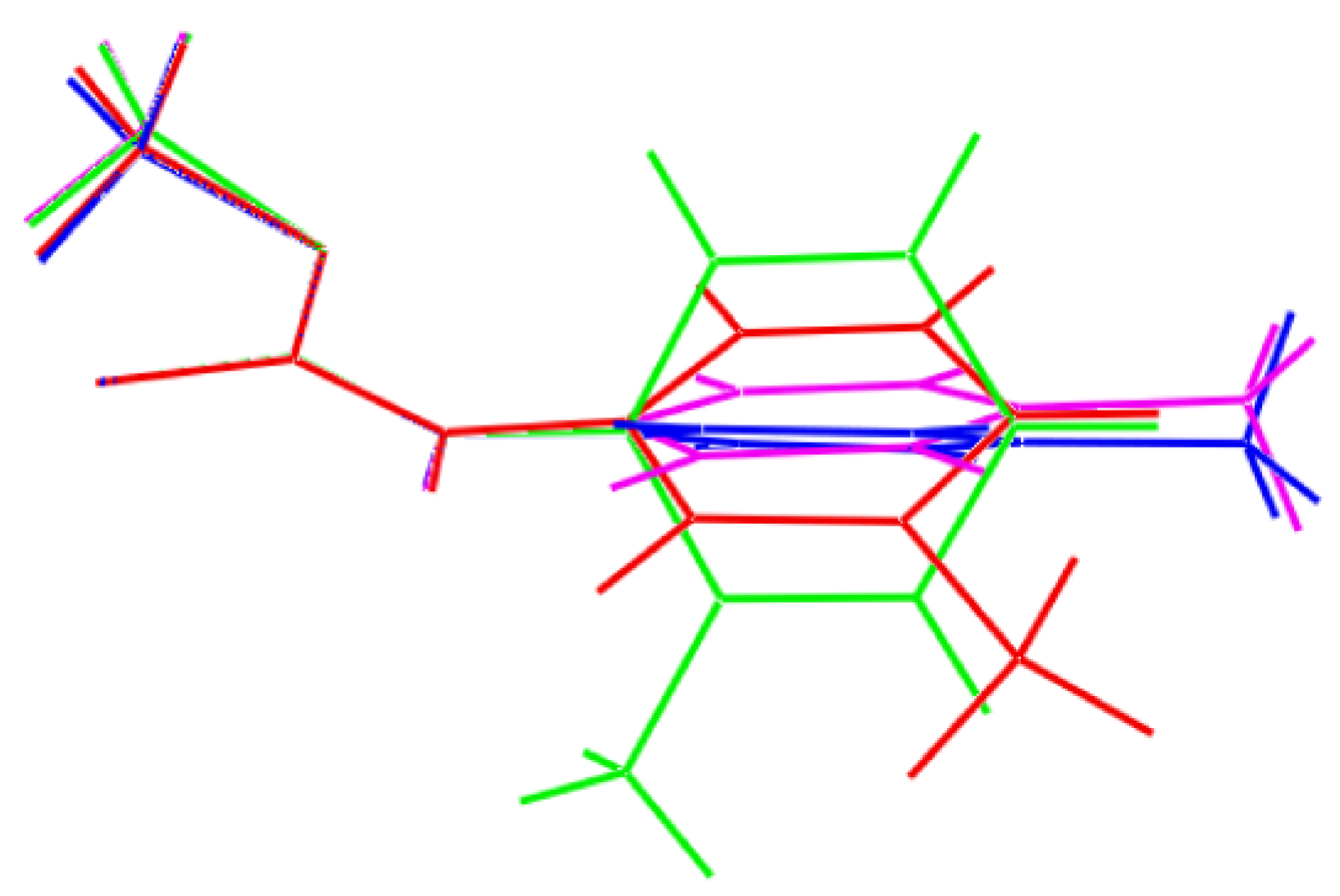
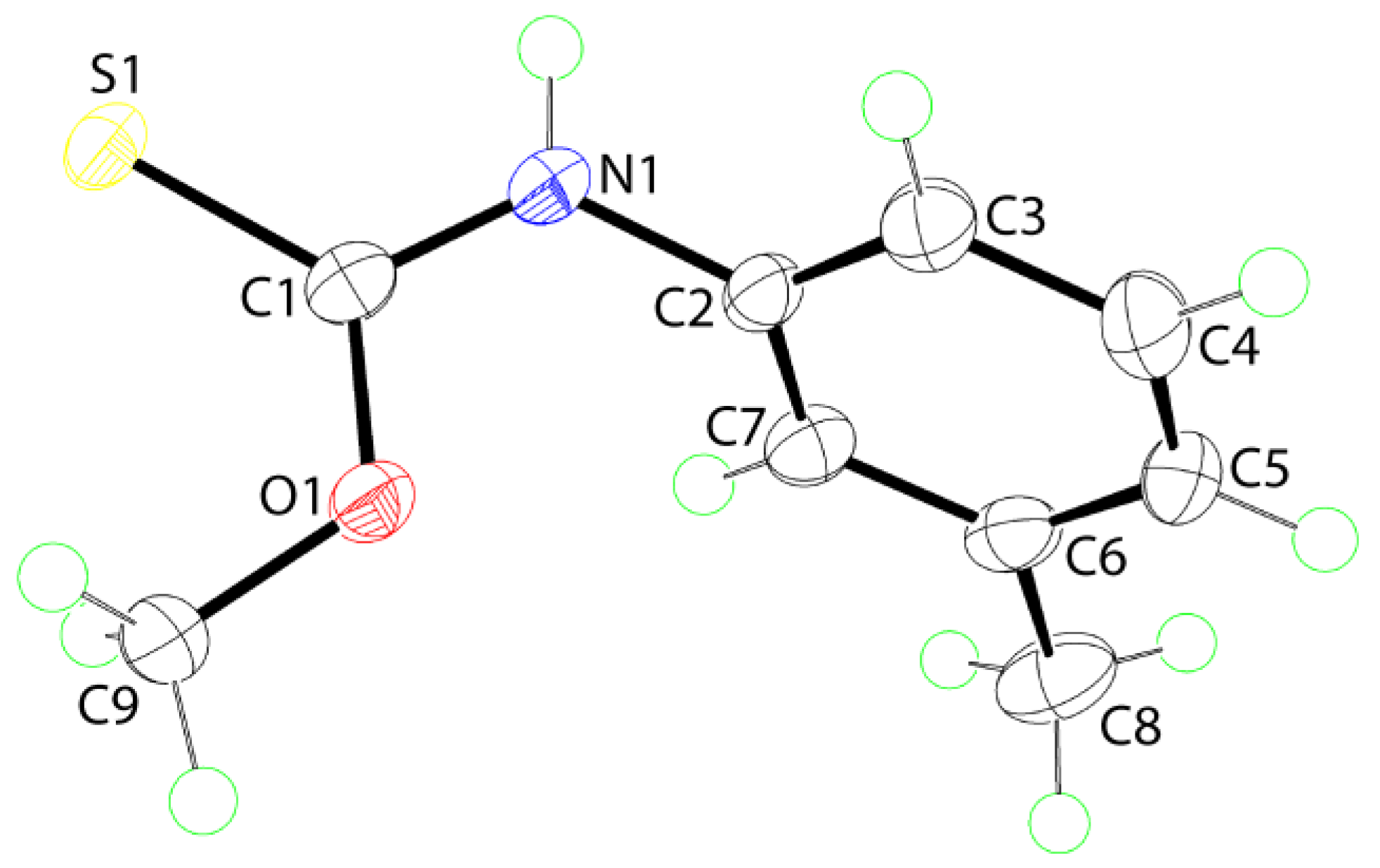
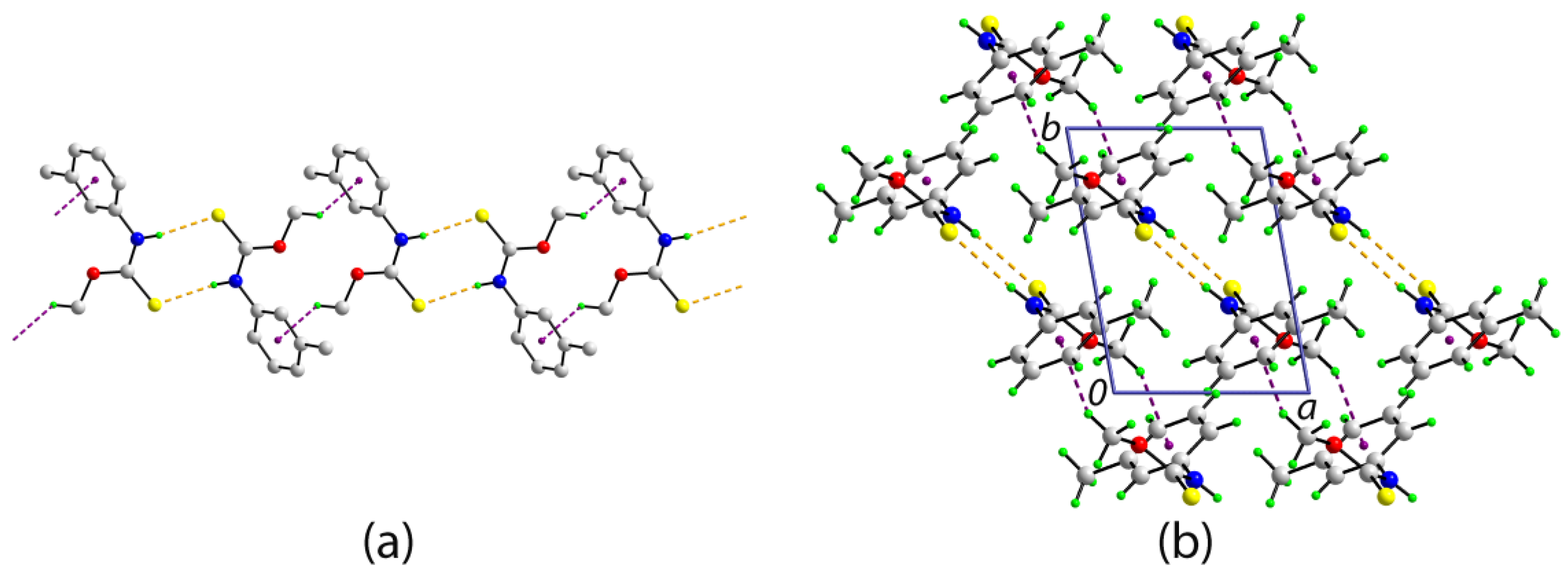
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Characterisation
3.3. Crystallography
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jotani, M.M.; Yeo, C.I.; Tiekink, E.R.T. A new monoclinic polymorph of N-(3-methylphenyl)ethoxycarbothioamide: Crystal structure and Hirshfeld surface analysis. Acta Crystallogr. E 2017, 73, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Bettens, R.P.A.; Dakternieks, D.; Duthie, A.; Tiekink, E.R.T. Prevalence of the thioamide {…H‒N‒C=S}2 synthon—Solid-state (X-ray crystallography), solution (NMR) and gas-phase (theoretical) structures of O-methyl-N-aryl-thiocarbamides. CrystEngComm 2005, 7, 682–689. [Google Scholar] [CrossRef]
- Xiao, H.-L.; Wang, K.-F.; Jian, F.-F. (4-Pyridyl)methyl N-phenylthiocarbamate. Acta Crystallogr. E 2006, 62, o2852–o2853. [Google Scholar] [CrossRef]
- Burrows, A.A.; Hunter, L. The associating effect of the hydrogen atom. Part XV*. The S-H-N bond. Esters of thion- and dithio-carbamic acids. J. Chem. Soc. 1952, 4118–4122. [Google Scholar] [CrossRef]
- Kuan, F.S.; Ho, S.Y.; Tadbuppa, P.P.; Tiekink, E.R.T. Electronic and steric control over Au…Au, C-H…O and C-H…interactions in the crystal structures of mononuclear triarylphosphinegold(I) carbonimidothioates: R3PAu[SC(OMe)=NR′] for R = Ph, o-tol, m-tol or p-tol, and R′ = Ph, o-tol, m-tol, p-tol or C6H4NO2-4. CrystEngComm 2008, 10, 548–564. [Google Scholar] [CrossRef]
- Ambinter. Available online: http://www.ambinter.com/ (accessed on 13 September 2018).
- FCH Group, Latvia. Available online: http://fchgroup.net/ (accessed on 13 September 2018).
- Ho, S.Y.; Cheng, E.C.C.; Tiekink, E.R.T.; Yam, V.W.W. Luminescent phosphine gold(I) thiolates: Correlation between crystal structure and photoluminescent properties in [R3PAu{SC(OMe)=NC6H4NO2-4}] (R = Et, Cy, Ph) and [(Ph2P-R-PPh2){AuSC(OMe)=NC6H4NO2-4}2] (R = CH2, (CH2)2, (CH2)3, (CH2)4, Fc). Inorg. Chem. 2006, 45, 8165–8174. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, F.S.; Tadbuppa, P.; Tiekink, E.R.T. Crystal structure of o-methyl N-(o-tolyl)thiocarbamate, SC(OCH3)NH(C6H4CH3). Z. Kristallogr. New Cryst. Struct. 2005, 220, 393–396. [Google Scholar]
- Ho, S.Y.; Kuan, F.S.; Tiekink, E.R.T. (E)-O-Methyl N-(4-methylphenyl)thiocarbamate. Acta Crystallogr. E 2007, 63, o1723–o1724. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction, CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2011.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, C.I.; Tiekink, E.R.T. O-Methyl m-Tolylcarbamothioate. Molbank 2018, 2018, M1020. https://doi.org/10.3390/M1020
Yeo CI, Tiekink ERT. O-Methyl m-Tolylcarbamothioate. Molbank. 2018; 2018(3):M1020. https://doi.org/10.3390/M1020
Chicago/Turabian StyleYeo, Chien Ing, and Edward R. T. Tiekink. 2018. "O-Methyl m-Tolylcarbamothioate" Molbank 2018, no. 3: M1020. https://doi.org/10.3390/M1020
APA StyleYeo, C. I., & Tiekink, E. R. T. (2018). O-Methyl m-Tolylcarbamothioate. Molbank, 2018(3), M1020. https://doi.org/10.3390/M1020