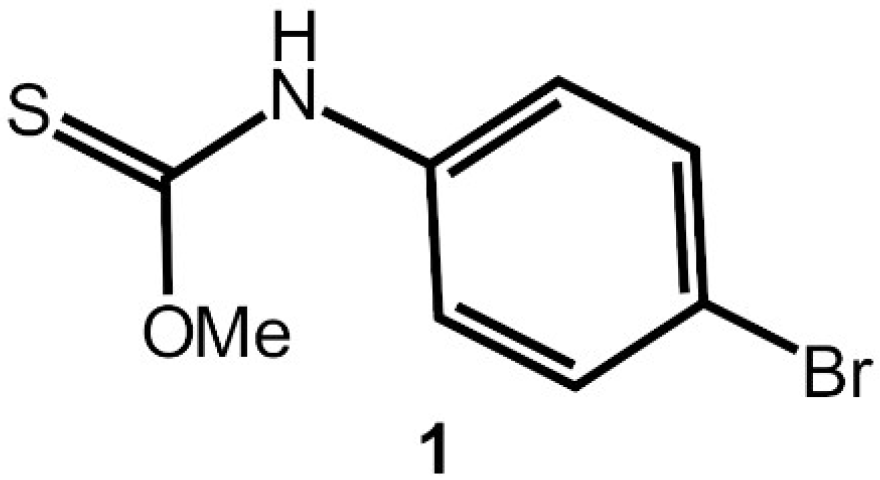
N-(4-Bromophenyl)methoxycarbothioamide



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
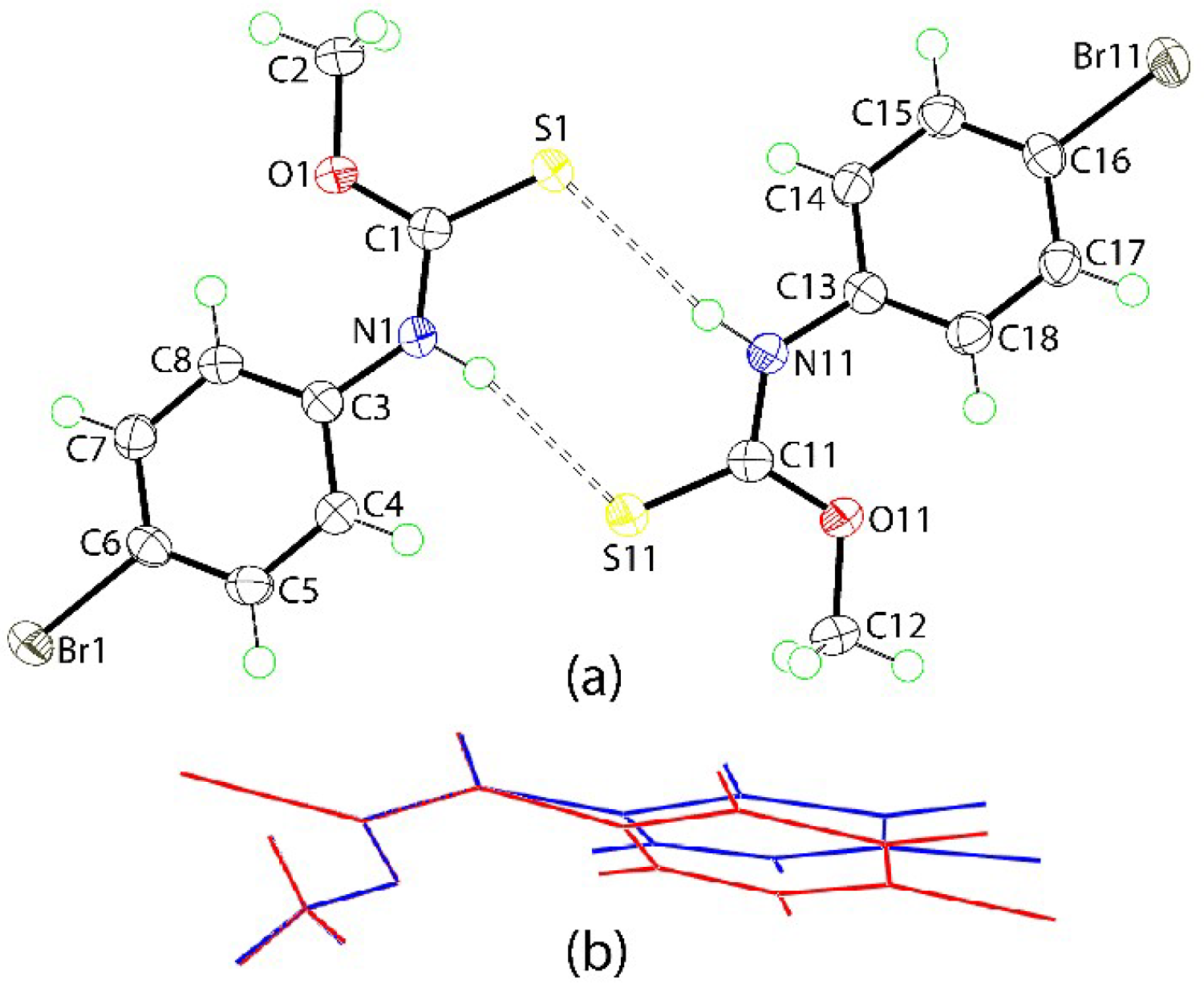
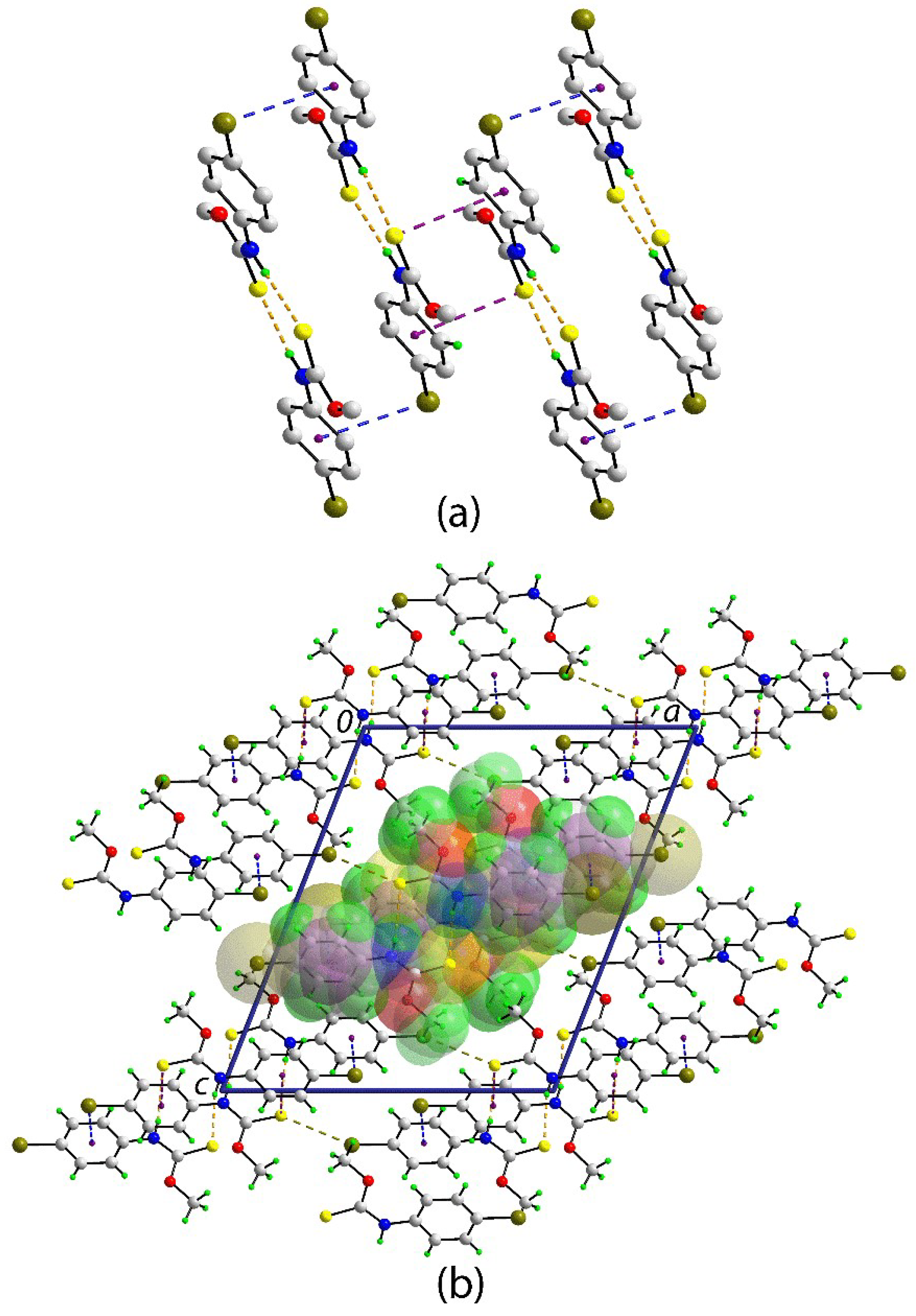
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Characterisation
3.3. Crystallography
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ooi, K.K.; Yeo, C.I.; Mahandaran, T.; Ang, K.P.; Akim, A.M.; Cheah, Y.K.; Seng, H.L.; Tiekink, E.R.T. G2/M cell cycle arrest on HT-29 cancer cells and toxicity assessment of triphenylphosphanegold(I) carbonimidothioates, Ph3PAu[SC(OR)=NPh], R = Me, Et, and iPr, during zebrafish development. J. Inorg. Biochem. 2017, 166, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Sim, J.H.; Khoo, C.H.; Goh, Z.J.; Ang, K.P.; Cheah, Y.K.; Fairuz, Z.A.; Halim, S.N.B.A.; Ng, S.W.; Seng, H.L.; et al. Pathogenic Gram-positive bacteria are highly sensitive to triphenylphosphanegold(O-alkylthiocarbamates), Ph3PAu[SC(OR)=N(p-tolyl)] (R = Me, Et and iPr). Gold Bull. 2013, 46, 145–152. [Google Scholar] [CrossRef]
- Ho, S.Y.; Cheng, E.C.C.; Tiekink, E.R.T.; Yam, V.W.W. Luminescent phosphine gold(I) thiolates: Correlation between crystal structure and photoluminescent properties in [R3PAu{SC(OMe)=NC6H4NO2-4}] (R = Et, Cy, Ph) and [(Ph2P-R-PPh2){AuSC(OMe)=NC6H4NO2-4}2] (R = CH2, (CH2)2, (CH2)3, (CH2)4, Fc). Inorg. Chem. 2006, 45, 8165–8174. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Khoo, C.H.; Chu, W.C.; Chen, B.J.; Chu, P.L.; Sim, J.H.; Cheah, Y.K.; Ahmad, J.; Halim, S.N.A.; Seng, H.L.; et al. The importance of Au…π(aryl) interactions in the formation of spherical aggregates in binuclear phosphane gold(I) complexes of a bipodal thiocarbamate dianion: A combined crystallographic and computational study, and anti-microbial activity. RSC Adv. 2015, 52, 41401–41411. [Google Scholar] [CrossRef]
- Ho, S.Y.; Bettens, R.P.A.; Dakternieks, D.; Duthie, A.; Tiekink, E.R.T. Prevalence of the thioamide {...H‒N‒C=S}2 synthon—solid-state (X-ray crystallography), solution (NMR) and gas-phase (theoretical) structures of O-methyl-N-aryl-thiocarbamides. CrystEngComm 2005, 7, 682–689. [Google Scholar] [CrossRef]
- Kuan, F.S.; Mohr, F.; Tadbuppa, P.P.; Tiekink, E.R.T. Principles of crystal packing in O-isopropyl-N-aryl-thiocarbamides: iPrOC(=S)N(H)C6H4-4-Y: Y = H, Cl, and Me. CrystEngComm 2007, 9, 574–581. [Google Scholar] [CrossRef]
- Jotani, M.M.; Yeo, C.I.; Tiekink, E.R.T. A new monoclinic polymorph of N-(3-methylphenyl)ethoxycarbothioamide: Crystal structure and Hirshfeld surface analysis. Acta Crystallogr. E 2017, 73, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.F.; Parken, E.R. 413. The unsaturation and tautomeric mobility of heterocyclic compounds. Part VI. The mobility of 5-substituted 1-hydroxybenzthiazoles, and the ultra-violet absorption of mobile and static derivatives of 1-hydroxybenzthiazole. J. Chem. Soc. 1935, 1755–1761. [Google Scholar] [CrossRef]
- Goeckeritz, D.; Pohloudek-Fabini, R. Halo and thiocyanato thiosemicarbazides and thiosemicarbazones and their behavior in paper chromatography. II. Pharmazie 1962, 17, 679–685. [Google Scholar]
- Goeckeritz, D.; Pohloudek-Fabini, R. Halo and thiocyanato thiourethans and their application to the determination of aliphatic alcohols by paper chromatography. Pharm. Zent. 1963, 102, 685–689. [Google Scholar]
- Bauman, R.A. Thioncarbamate esters. U.S. Patent US 3592835 A 19710713, 1971. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Jones, W.; Motherwell, W.D.S.; Shields, G.P. Crystal engineering and chloro-methyl interchange—A CSD analysis. Mol. Cryst. Liq. Cryst. 2001, 356, 337–353. [Google Scholar] [CrossRef]
- Ho, S.Y.; Lai, C.S.; Tiekink, E.R.T. O-Methyl N-phenylthiocarbamate. Acta Crystallogr. E 2003, 59, o1155–o1156. [Google Scholar] [CrossRef]
- Ho, S.Y.; Lai, C.S.; Tiekink, E.R.T. (E)-O-Methyl N-(4-methylphenyl)thiocarbamate. Acta Crystallogr. E 2007, 63, o1723–o1724. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
Sample Availability: Samples of the compound is not available from the authors. |



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, C.I.; Tiekink, E.R.T. N-(4-Bromophenyl)methoxycarbothioamide. Molbank 2018, 2018, M1012. https://doi.org/10.3390/M1012
Yeo CI, Tiekink ERT. N-(4-Bromophenyl)methoxycarbothioamide. Molbank. 2018; 2018(3):M1012. https://doi.org/10.3390/M1012
Chicago/Turabian StyleYeo, Chien Ing, and Edward R.T. Tiekink. 2018. "N-(4-Bromophenyl)methoxycarbothioamide" Molbank 2018, no. 3: M1012. https://doi.org/10.3390/M1012
APA StyleYeo, C. I., & Tiekink, E. R. T. (2018). N-(4-Bromophenyl)methoxycarbothioamide. Molbank, 2018(3), M1012. https://doi.org/10.3390/M1012