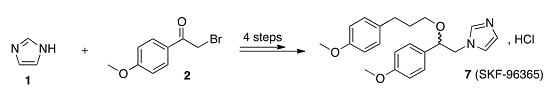
1. Introduction
Variations in cytoplasmic Ca
2+ concentration ([Ca
2+]
i) play an important role in mediating fundamental biological activities for cell proliferation, differentiation, apoptotosis, and gene expression. Calcium influx through store-operated Ca
2+ entry (SOCE) triggered by the depletion of endoplasmic reticulum (ER) Ca
2+ stores largely contributes to changes in ([Ca
2+]
i) and regulates a large variety of cellular functions [
1,
2,
3]. The first identified inhibitor of the CRAC (Ca
2+ release-activated Ca
2+ channel) channel, the archetypal SOCE channel, was the 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1
H-imidazole hydrochloride, also known as SKF-96365 [
4], and this imidazole derivative inhibited thapsigargin-induced SOCE in Jurkat cells with an IC
50 of 12 μM in a dose-dependant manner [
5]. Although this inhibitor, in a mouse model of breast cancer, prevented the development of tumor metathesis [
6]. In the imidazole group, econazole and miconazole have been also identified as inhibitors of CRAC channels, but some studies mentioned that these imidazole coumpounds showed non-specific effects by inhibiting the cytochrome P450 activity [
7] or other Ca
2+ channels [
8]. However, SKF-96365 is still used as a tool for the probing of receptor-mediated Ca
2+ entry processes, particularly in non-excitable cells [
9,
10,
11]. To our surprise, the synthesis of SKF-96365 was not patented or not detailed in academic literature, but it is interesting to note that the enantiomers has been subjected to a chiral high pressure liquid chromatography (HPLC) study [
12]. In this context and due to the recent interest of SKF-96365 for their anticancer effect in a mouse xenograph model [
13], we developed efforts on a complete and detailed preparation of 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1
H-imidazole hydrochloride.
2. Results
The synthetic route for the preparation of the title compound
7 was achieved in four steps, as presented in
Scheme 1. In the first step, reaction of commercial imidazole
1 with 2-bromo-4′-methoxyacetophenone
2 produced mainly the corresponding 2-(1
H-imidazol-1-yl)-1-[4-methoxyphenyl)ethanone
3 in a 51% yield. This
N-alkylation was conducted in DMF at 5 °C and after a reaction time of two hours; the crude reaction mixture was treated with deionized water in order to eliminate the undesired
N-protoned derivative
3.
For the second step, the acetophenone derivative
3 in a dry methanol solution was converted into its corresponding alcohol
4 [
14] in a 63% yield by the addition of sodium borohydride (2 equiv.). In the third step, treatment of alcohol
3 with 4-methoxyphenylpropylbromide
5 in a basic (KOH, 5 equiv.) solution of dimethylsulfoxide led to the desired
O-alkyl compound
6 together with the
N-alkyl derivative of
6 as a by-product. Compound
6 was purified by preparative chromatography on silica gel using a step-wise gradient of CH
2Cl
2/MeOH (0–1%). Finally, from a solution of 1M HCl in diethyl ether, pure
O-alkyl imidazole intermediate
6 was transformed into imidazolium chloride
7. It is interesting to note that the crystallization was achieved after one day at 25 °C in a 29% yield. Compound
7 was prepared with an overall yield of 9%. The structure of
7 was confirmed by
1H-NMR, and the data were in agreement with those previously described in literature [
12].
13C-NMR, HRMS, and elemental analysis of
7 completed the data and are in accordance with this structure.
3. Experimental Section
3.1. General Information
Preparative chromatography was realized on a Combi Flash Rf 200 psi UV ref. 208K20284 (Serlabo Technologies, Entraigues-sur-la-Sorgue, France) using pre-packed column of alumina gel 60 F 254 Merck equipped with a DAD UV/Vis 200–360-nm detector . Thin-layer chromatography (TLC) was accomplished on 0.2-mm precoated plates of neutral alumina gel 60 F-254 (Merck) with appropriate eluent. Visualization was made with ultraviolet light (254 and 365 nm) or with a fluorescence indicator. Solvents were evaporated with a BUCHI rotary evaporator. All reagents and solvents were purchased from Acros Fisher, Sigma-Aldrich Chimie, and Fluka France and were used without further purification. 1H-NMR spectra were recorded on a BRUKER AC 300 P (300 MHz) spectrometer, and 13C-NMR spectra on BRUKER AC 300 P (75 MHz) spectrometer (Bruker France Scientifique, Voisins-le-Bretonneux, France). Chemical shifts are expressed in parts per million downfield. Data are given in the following order: δ value, multiplicity (s: singlet; d: doublet; t: triplet; q: quartet; quint: quintuplet: m: multiplet; br: broad), number of protons, coupling constants J is given in Hertz. The high-resolution mass spectra (HRMS) were recorded in positive mode using direct electrospray infusion, respectively on Waters Q-Tof 2 or on Thermo Fisher Scientific Q-Exactive spectrometers, and the purity of the final compound 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1H-imidazole hydrochloride 7 was performed on a microanalyzer Flash EA1112 CHNS/O Thermo Electron (Thermo Electron, Villebon-sur-Yvette, France). The obtained results were within ±0.4% of the theorical values at the "Centre Régional de Mesures Physiques de l’Ouest" SFS ScanMAT (CRMPO SFS ScanMAT, Rennes, France). Melting points were determined on a Kofler melting point apparatus and were uncorrected.
3.2. Synthesis of 2-(1H-Imidazol-1-Yl)-1-[4-Methoxyphenyl]Ethanone (3)
In a 50-mL round-bottomed flask provided with a magnetic stirrer and condenser containing a suspension of commercial imidazole (1, 3.09 g, 45.4 mmol, 5.2 equiv.) in DMF (2.16 mL) cooled at 5 °C (with an ice bath), small portions of 2-bromo-4′-methoxyacetophenone (2, 2 g, 8.73 mmol, 1 equiv.) were added over a period of 15 min. The resulting suspension was stirred vigorously (500 rpm) for 2 h at a temperature below 15 °C. The reaction mixture was monitored by thin-layer chromatography on 0.2-mm pre-coated plates of silica gel 60F-254 (Merck) using a mixture of CH2Cl2/MeOH (9:1 v/v) as eluent. Deionized water (60 mL) was added to the heterogeneous mixture, and the flask was manually swirled twice for 5 min to homogenize; then, the resulting mixture was maintained at 5 °C (refrigerator) without stirring for 12 h to improve precipitation and decantation. The resulting precipitate in the reaction mixture was collected by filtration on a Büchner funnel (porosity N°4) and solubilized in methylene chloride (44 mL). The organic phase was filtered on filter paper and transferred into a separating funnel. The organic phase was washed with saturated sodium bicarbonate (4 × 44 mL) and dried over magnesium sulfate. The filtrate was concentrated in a rotary evaporator under reduced pressure and finally dried in a high vacuum (10−2 Torr) at 25 °C for 2 h. The desired compound 3 was obtained in a 51% yield as a beige powder and was further used without purification. Mp = 135–137 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.87 (s, 3H, OCH3), 5.66 (s, 2H, CH2, H-2), 6.91 (t, 1H, J = 1.1 Hz, H-5′, Ar), 7.10 (t, 1H, J = 1.2 Hz, H-4′, Ar), 7.11 (d, 2H, J = 8.9 Hz, H-3′′, H-5′′), 7.57 (t, 1H, J = 1.1 Hz, H-2′, Ar), 8.02 (d, 2H, J = 8.9 Hz, H-2′′, H-6′′, Ar). 13C-NMR (75 MHz, DMSO-d6) δ 52.2 (OCH3), 55.7 (C-2), 114.2 (C-3′′, C-5′′), 121.0 (C-5′), 127.3 (C-1′′), 127.8 (C-4′), 130.4 (C-2′′, C-6′′), 138.4 (C-2′), 163.7 (C-4′′), 191.9 (C-1). ES+ HRMS, m/z = 216.0790 found (calculated for C12H12N2O2Na [M + Na]+ requires 216.0971).
3.3. Synthesis of 2-(1H-Imidazol-1-Yl)-1-[4-Methoxyphenyl]Ethanol (4)
In a 100-mL round-bottomed flask provided with a magnetic stirrer and condenser, 2-(1H-imidazol-1-yl)-1-[4-methoxyphenyl]ethanone (3, 1.046 g, 4.84 mmol, 1 equiv.) was solubilized in methanol for HPLC (41.8 mL) at 0 °C (in ice bath) under vigorous magnetic stirring (550 rpm). Commercial sodium borohydride (366 mg, 9.67 mmol, 2 equiv) was added to this homogeneous solution in small portions over a period of 10 min. The resulting mixture was stirred at room temperature for 2 h and monitored by thin-layer chromatography on 0.2 mm pre-coated plates of silica gel 60F-254 (Merck) using a mixture of CH2Cl2/MeOH (95:5 v/v) as eluent. Then, the reaction mixture was concentrated in a rotary evaporator under reduced pressure, and the crude residue was dissolved in deionized water (43.5 mL). This solution was transferred into a separating funnel and extracted with methylene chloride (3 × 43.5 mL). The combined organic phases were dried over magnesium sulphate and filtered on filter paper, and the solvent of the filtrate was eliminated in vacuo. The desired compound 4 was obtained in a 63% yield as a white powder and further used without purification. Mp = 167–169 °C. 1H-NMR (300 MHz, DMSO-d6) δ 3.74 (s, 3H, OCH3), 4.00 (dd, 1H, J = 7.6, 13.8 Hz, CH, H-2), 4.08 (dd, 1H, J = 4.4, 13.8 Hz, CH, H-2), 4.66–4.80 (dt, 1H, J = 4.4, 8.4 Hz, CH, H-1), 5.56 (br d, 1H, J = 4.5 Hz, OH), 6.81 (t, 1H, J = 1.0 Hz, H-5′, Ar), 6.84–6.92 (d, 2H, J = 1.1 Hz, H-3′′, H-5′′, Ar), 7.09 (t, 1H, J = 1.2 Hz, H-4′, Ar), 7.20–7.27 (m, 2H, H-2′′, H-6′′, Ar), 7.47 (t, 1H, J = 1.1 Hz, H-2′, Ar). 13C-NMR (75 MHz, DMSO-d6) δ 53.6 (C-2), 55.0 (OCH3), 71.6 (C-1), 113.4 (C-3′′, C-5′′), 120.0 (C-5′), 127.2 (C-2′′, C-6′′), 127.7 (C-4′), 134.6 (C-1′′), 137.6 (C-2′), 158.5 (C-4′′). ES+ HRMS, m/z = 249.0949 found (calculated for C12H14N2O2Na [M + Na]+ requires 241.0948).
3.4. Synthesis of 1-{β-[3-(4-Methoxy-Phenyl)Propoxy]-4-Methoxyphenethyl}-1H-Imidazole Hydrochloride (7)
A portion of 2-(1H-imidazol-1-yl)-1-[4-methoxyphenyl]ethanol (4, 790 mg, 3.62 mmol, 1 equiv.), pellets of potassium hydroxide (1.015 g, 18.1 mmol, 5 equiv.), and dry dimethylsulfoxide (13.7 mL) were placed successively in a 10-mL two-necked round-bottomed flask (provided with a magnetic stirrer and condenser) under a stream of argon. This mixture was stirred (500 rpm) at room temperature for 1 h, and one portion of 1-(3-bromoproyl)-4-methoxy benzene (5, 0.63 mL, 3.62 mmol, 1 equiv.) was added. The reaction mixture was heated at 60 °C for 48 h under vigorous magnetic stirring (700 rpm) under an argon atmosphere and monitored by thin-layer chromatography on 0.2-mm pre-coated plates of silica gel 60F-254 (Merck) using a mixture of CH2Cl2/MeOH (9:1 v/v) as eluent. After cooling down to room temperature, saturated brine (79 mL) was poured into the reaction mixture. The resulting solution was transferred into a separating funnel and extracted twice with diethyl ether (39.5 mL). After decantation, the organic phase was washed three times with water (79 mL), dried over magnesium sulphate, and filtered through filter paper. The solvent of the filtrate was eliminated in a rotary evaporator. The oily residue (crude 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1H-imidazole 6) was submitted to purification by preparative chromatography (Combi Flash Rf 200 psi apparatus with a DAD 200/360 nm detector) on a pre-packed column of silica gel 60F 254 (Merck) using a stepwise gradient of methylene chloride/methanol (0–1%). Pooling (flow rate: 18 mL/min during 60 min, retention time: 45 min) and the elimination of the solvent in vacuo gave the pure desired intermediate 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1H-imidazole (6, 470 mg, 1.28 mmol) as a mobile pale orange oil.
The oily compound 6 was dissolved in dry ether (6.6 mL) under a stream of argon with magnetic stirring (200 rpm). A solution of 1M HCl in ether (1.28 mL, 1.28 mmol) was poured into the homogeneous solution. During mixing at 25 °C, the initial pale orange solution began to crystallize progressively on the circumference of the round flask after 15 min, and mixing was pursued until the complete precipitation of the 1-{β-[3-(4-methoxy-phenyl)propoxy]-4-methoxyphenethyl}-1H-imidazole hydrochloride 7 (after 12 h at 25 °C). The resulting precipitate was carefully triturated in the flask with ether to obtain a divided powder, and this slurry was mixed vigorously (600 rpm) for 12 h. The insoluble salt was finally collected in a Büchner funnel (porosity N°4), was washed with dry ether (3 × 10 mL), and was dried under a high vacuum (10−2 Torr) at 25 °C for 8 h. 1H, 13C-NMR, HRMS, and centesimal analysis characterized the hydrochloride salt 7 obtained in a 29% yield as a white powder. Mp = 117.5–119.5 °C. 1H-NMR (300 MHz, MeOH-d4) δ 1.77 (tt, J = 6.2, 7.5 Hz, 2H, CH2, H-2a), 2.51 (dd, J = 6.6, 8.4 Hz, 2H, CH2, H-3a), 3.22 (dt, J = 6.1, 9.4 Hz, 1H, CH, H-1a), 3.36 (dt, J = 6.2, 9.4 Hz, 1H, CH, H-1a), 3.74 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 4.40–4.48 (m, 2H, CH2, H-2), 4.64 (dd, J = 5.2, 6.6 Hz, 1H, CH, H-1), 6.78 (d, J = 8.6 Hz, 2H, H-2b, H-6b, Ar), 6.90–7.04 (m, 4H, H-3′′, H-5′′, H-3b, H-5b, Ar), 7.25 (d, J = 8.7 Hz, 2H, H-2′′, H-6′′, Ar), 7.56 (t, J = 1.7 Hz, 1H, H-5′, Ar), 7.62 (t, J = 1.7 Hz, 1H, H-4′, Ar), 8.84 (t, J = 1.7 Hz, 1H, H-2′, Ar).13C-NMR (75 MHz, MeOH-d4) δ 32.3 (C-2a), 32.6 (C-3a), 55.7 (OCH3), 55.8 (OCH3), 56.0 (C-2), 69.1 (C-1a), 80.6 (C-1), 114.8 (C-2b, C-6b), 115.3 (C-3b, C-5b), 120.6 (C-5′), 124.3 (C-4′), 129.1 (C-2′′, C-6′′), 130.3 (C-3′′, C-5′′), 130.6 (C-1b), 134.9 (C-1′′), 137.3 (C-2′Ζ), 159.4 (C-4b), 161.6 (C-4′′). ES+ HRMS, m/z = 367.2015 found (calculated for C22H27N2O3 [M + H]+ requires 367.2016 and 389.1829 found (calculated for C22H26N2O3Na [M + Na]+ requires 389.1836). Anal. calcd. for C22H27ClN2O3 (402.91): C, 65.58; H, 6.75; N, 6.95. Found: C, 65.48; H, 6.56; N, 6.75.


 ,
, 
{kind=link}
{kind=link}
