Introduction
In nature steroid dimers are synthesized by some marine organisms, but, due to their interesting pharmacological properties, various routes to synthetic derivatives have also been developed [
1,
2,
3].
Among natural steroid dimers, cephalostatines and ritterazines have significant antitumor activity [
4]. Synthetic steroid dimers may have micellar [
5], detergent or liquid crystal properties or can be used as catalysts in some organic reactions [
6]. They can also be applied in drug delivery [
3] or in supramolecular chemistry [
7]. The biological activity of some dimeric steroid esters showed considerable prolongation of hormonal effects compared to the respective monomer [
8]. Other derivatives were found to show appreciable cytotoxicity [
9].
Pregnenolone (3β-hydroxypregn-5-en-20-one) is involved in several pathophysiological events, such as response to stress, depression, anxiety, sleep, epilepsy, and memory formation [
10]. Its metabolite, pregnenolone sulfate modulates a variety of ion channels, transporters, and enzymes [
11].
Consequently, among new steroid dimers, a number of pregnenolone derivatives were synthesized for pharmacological screening [
12,
13].
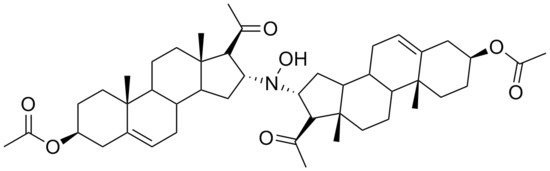
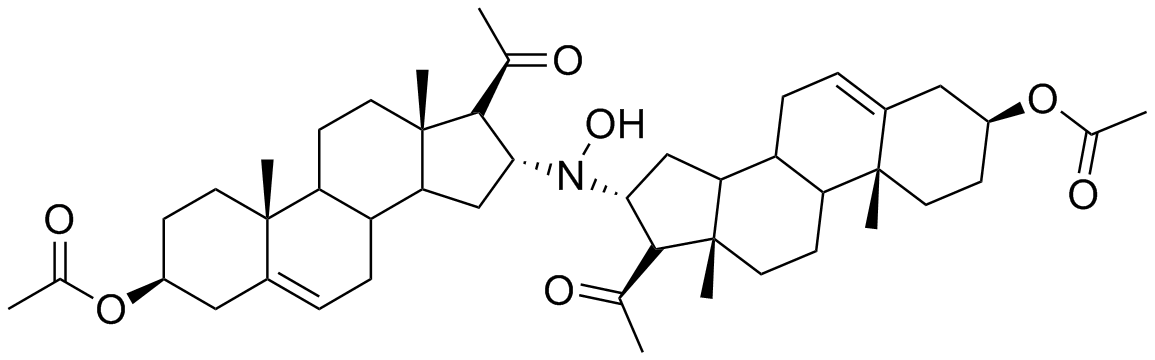
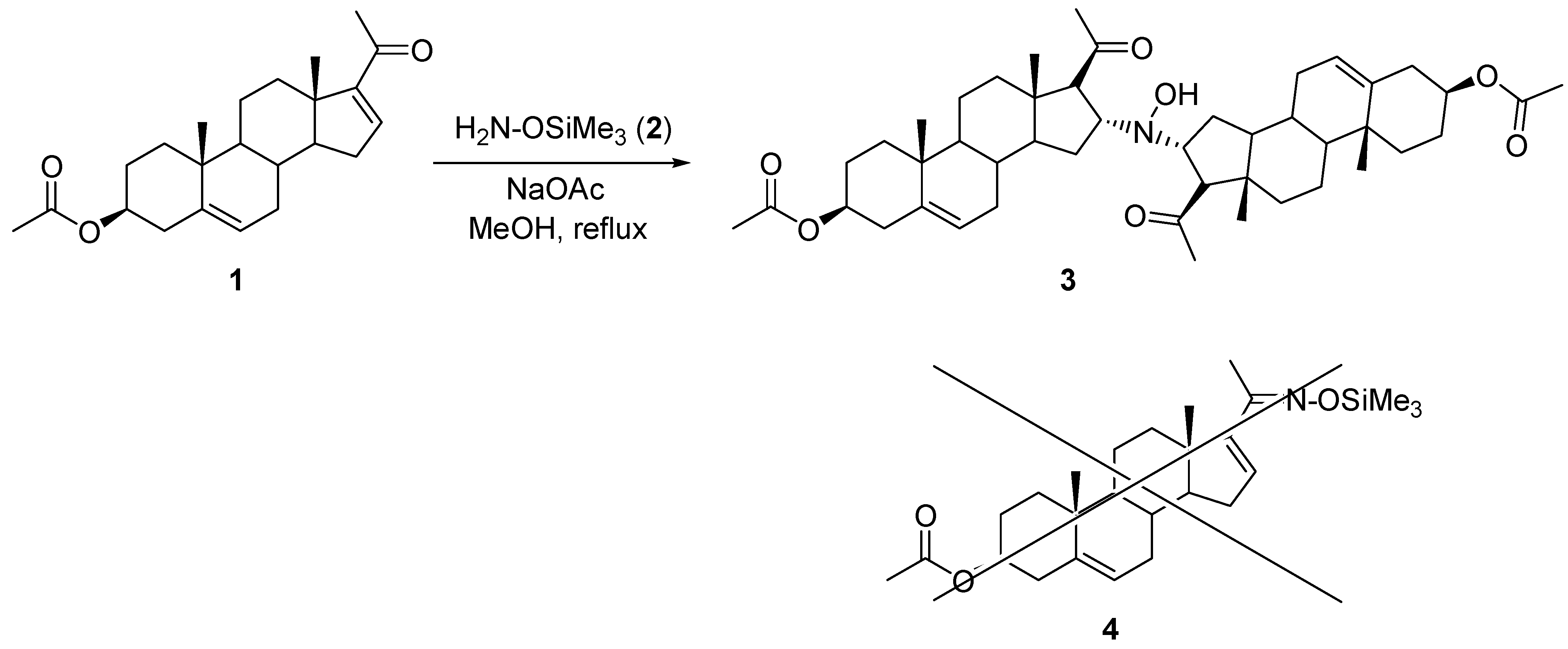
During our research concerning the synthesis of new steroid derivatives, we have found that the dimeric derivative
N,
N-bis(3β-acetoxypregn-5(6)-en-20-on-16α-yl)hydroxylamine (
3,
Scheme 1) could be obtained in the base catalyzed reaction of 3β-acetoxypregn-5,16-dien-20-one (
1) with
O-(trimethylsilyl)hydroxylamine (
2).
Results and Discussion
Upon reacting 3β-acetoxypregn-5,16-dien-20-one (
1,
Scheme 1) with
O-(trimethylsilyl)hydroxylamine (
2) in the presence of sodium acetate in refluxing methanol, a white precipitate was formed. Spectroscopic analysis of this material did not support the formation of the expected oxime derivative (
4), so a detailed study including
1H and
13C-NMR measurements together with 2D experiments (COSY, HSQC, HMBC, ROESY), as well as IR spectroscopy, FAB MS and elemental analysis, was carried out to elucidate the structure of the product.
The FAB MS with an [M+H]+ signal of m/z = 746 strongly supported the formation of a dimeric derivative. The presence of only two singlets of angular methyl groups in the 1H-NMR at 0.99 ppm and 0.63 ppm suggested a symmetric structure.
In the
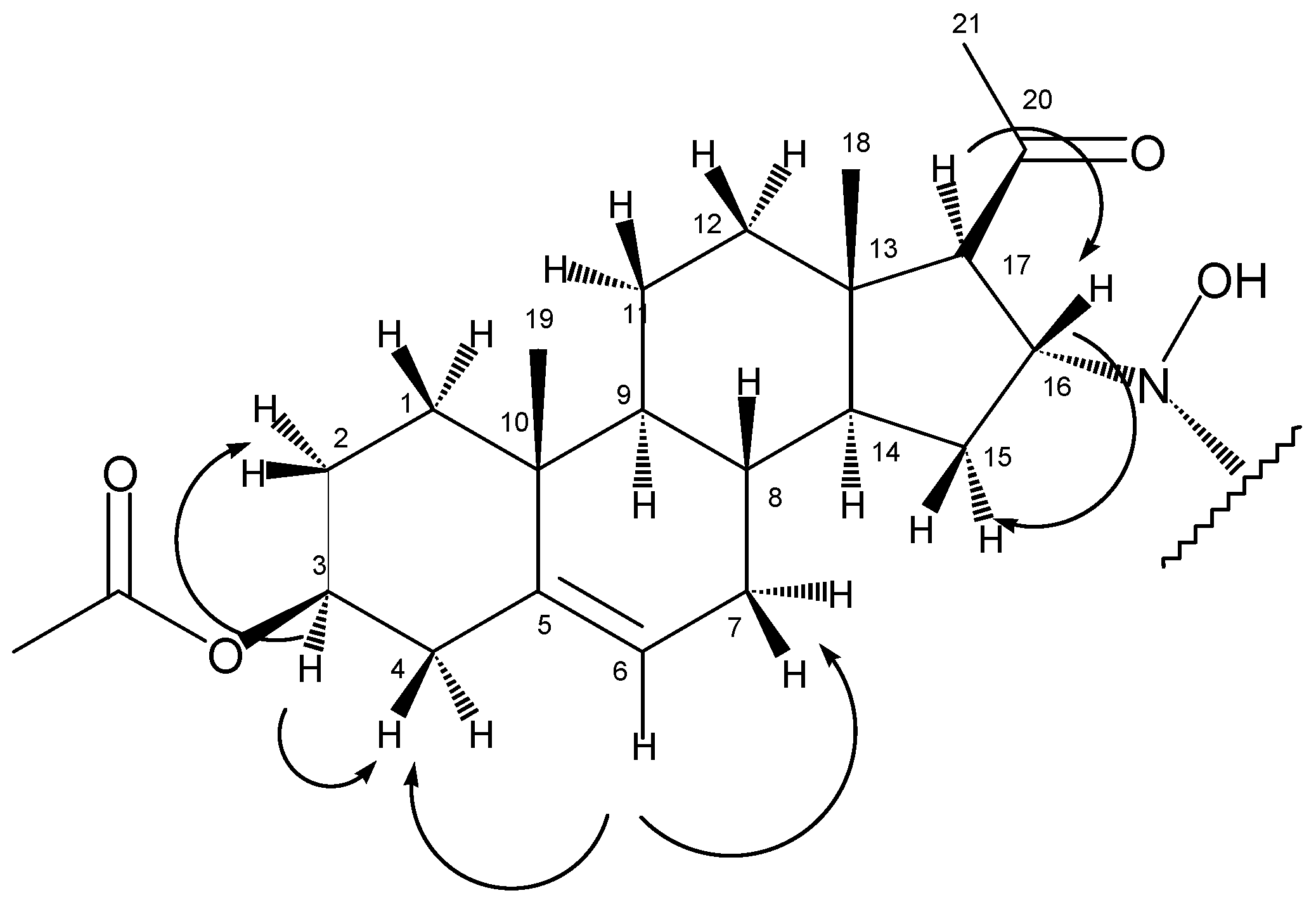
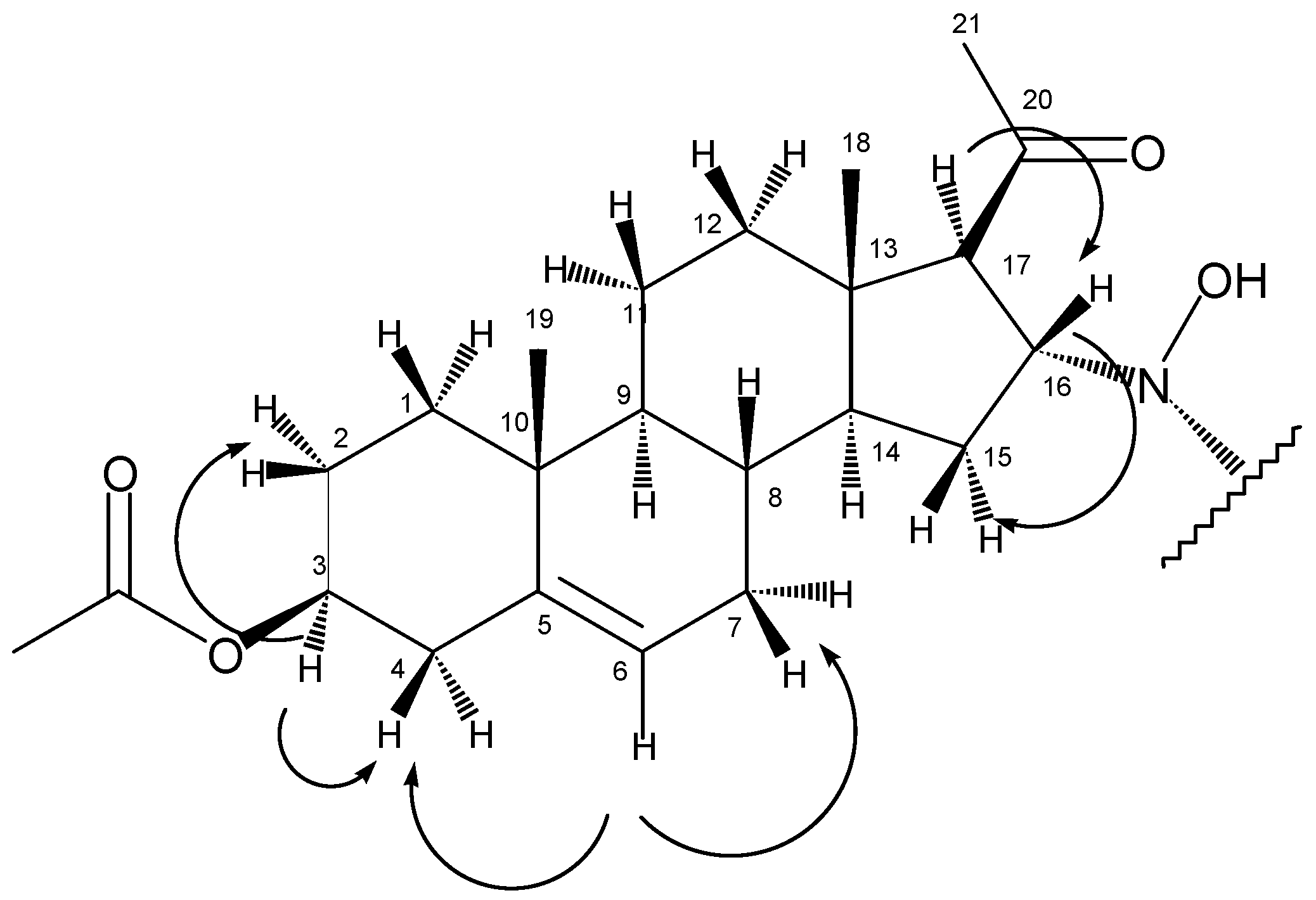
1H-NMR spectrum, beside the peaks at 5.35 ppm (6-H) and 4.58 ppm (3-H) that correspond well to the chemical shifts of the same protons of the starting material, the appearance of a multiplet at 3.96 ppm and a doublet at 2.77 ppm could be observed. These peaks, that were proved to be coupled nuclei based on the COSY experiment (
Figure 1), together with the lack of the signal of an olefinic proton around 6 ppm, support an addition reaction on the Δ
16 double bond of the steroidal substrate
1.
The COSY experiment showed that in accordance with the splitting pattern of the peak at 2.77 ppm, this proton had only one coupling partner at 3.96 ppm, while the latter signal gave crosspeaks with methylene protons with chemical shifts of 1.32 ppm and 1.85 ppm. These observations helped to assign the four protons as follows: 17-H at 2.77 ppm, 16-H at 3.96 ppm, 15-H2 at 1.32 ppm and 1.85 ppm. From the HSQC experiment, the signals of the corresponding carbon atoms could also be deduced (C17: 68.0 ppm, C16: 65.8 ppm, C15: 36.7 ppm).
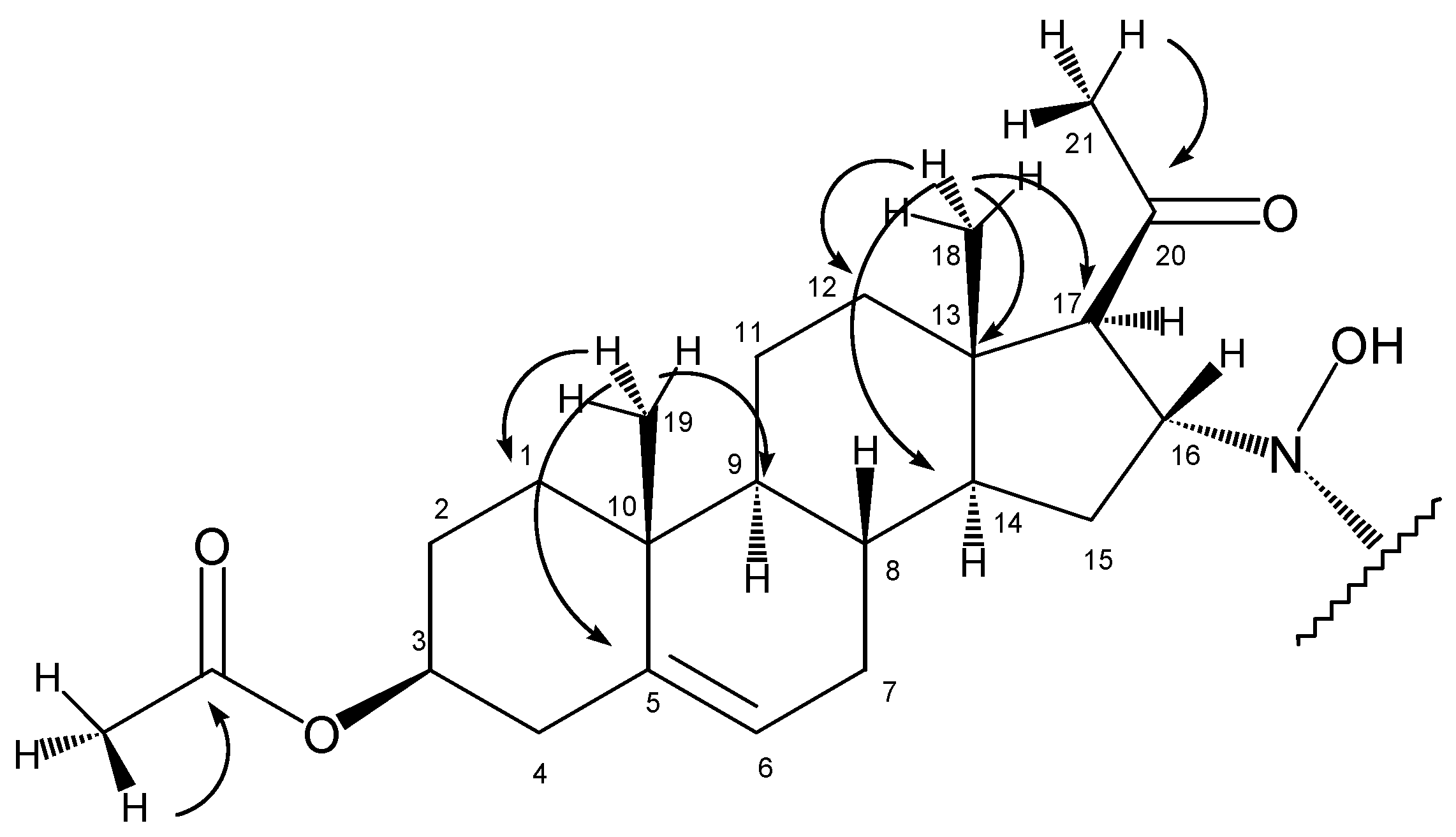
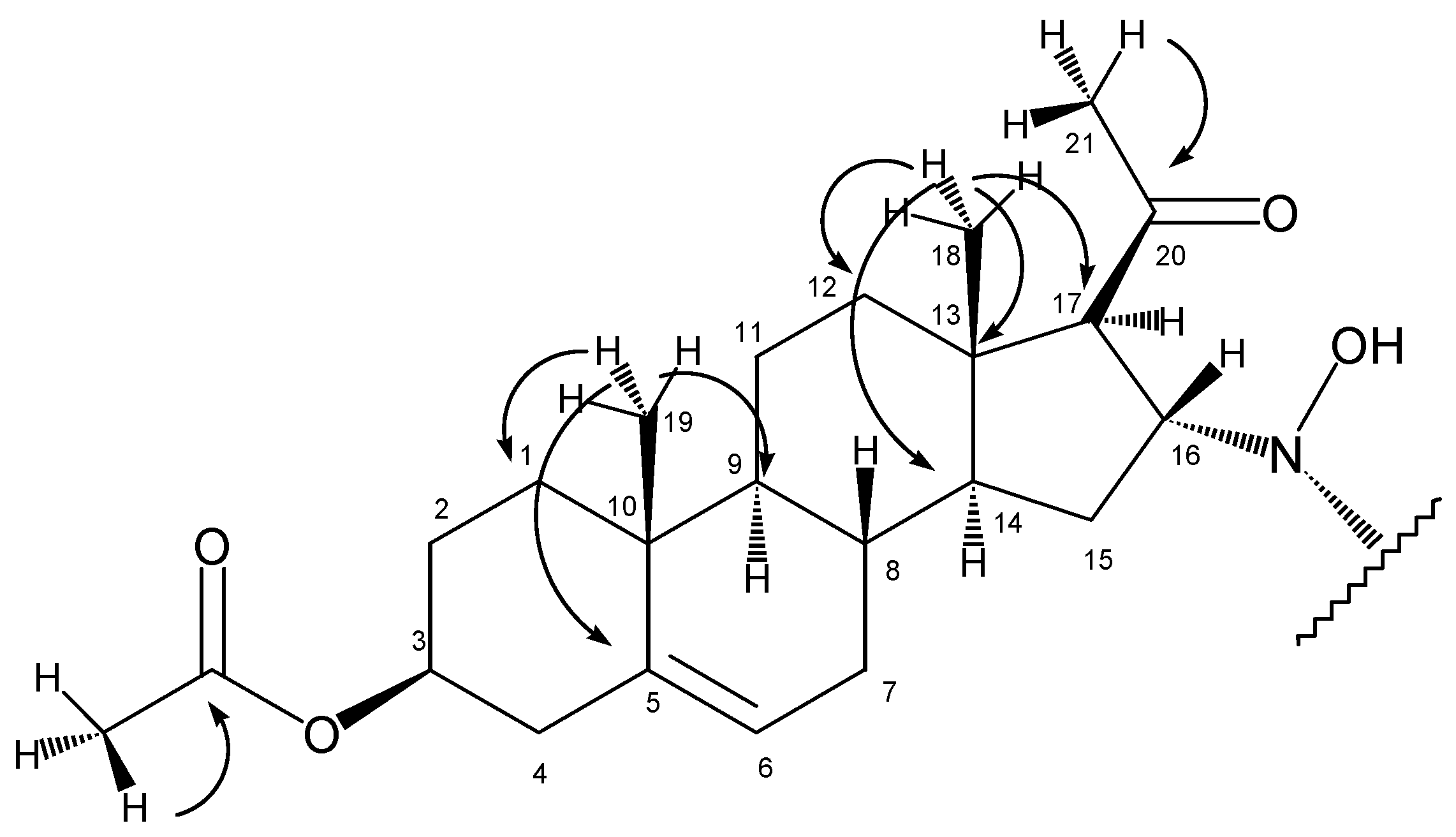
In the HMBC spectrum, the olefinic carbon C5 (at 139.3 ppm) gave a crosspeak with the methyl singlet at 0.99 ppm (
Figure 2), so the latter could be assigned as 19-H
3. From this information, chemical shifts of C19 (19.0 ppm), 1-CH
2 (1-H
2: 1.10 ppm and 1.80 ppm, C1:36.4 ppm) and 9-CH (9-H: 1.00 ppm, C9: 49.5 ppm) could be determined with the help of HSQC and HMBC experiments.
According to the HSQC spectrum, the proton giving the multiplet at 5.35 ppm is bonded to the carbon with the signal of 122.1 ppm, so these peaks could be attributed to 6-CH. Based on the COSY results, 6-H is coupled to two methylene groups with chemical shifts of 1.62 ppm, 2.00 ppm and 2.30 ppm (the latter with two protons of identical chemical shift). The latter signal could be assigned to 4-H2 based on its crosspeak with 3-H at 4.58 ppm. As a consequence, the former two peaks belonged to 7-H2. Other coupling partners of 3-H were found at 1.50 and 1.85 ppm, so these could be defined as 2-H2. The corresponding carbon signals are as follows: C7 at 31.4 ppm, C4 at 37.8 ppm, C3 at 73.6 ppm and C2 at 22.5 ppm.
Starting from the other methyl singlet at 0.63 ppm (18-H3), chemical shifts of C18 (14.1 ppm), C14 (55.0 ppm), C13 (44.0 ppm), C12 (38.4 ppm), 14-H (1.32 ppm) and 12-H2 (1.50 ppm and 1.90 ppm) could be determined using the HSQC and HMBC spectra.
The two methyl singlets at 2.12 ppm and 2.01 ppm are the signals of 21-H3 and protons of the acetoxy group on C3. These assignments were based on their crosspeaks with carbon signals at 208.1 ppm (carbonyl carbon) and 170.3 ppm (ester CO), respectively, in the HMBC spectrum.
The stereochemistry of C17 and C16 was determined with the help of a ROESY experiment showing the proximity of 18-H3 and 16-H. This observation proved the β disposition of 16-H, while the lack of a signal between either 16-H and 17-H or 18-H3 and 17-H supported the α disposition of 17-H.
The lack of a signal corresponding to SiMe3 in the 1H-NMR and the appearance of the broad band of OH stretching around 3500 cm−1 in the IR spectrum supported the hydrolysis of the trimethylsilyl group.
It should be mentioned however, that the reactions of steroid 1 with hydroxylamine or O-methylhydroxylamine led to the corresponding oximes under the same conditions. The first observation supports the assumption that cleavage of the trimethylsilyl group takes place after the nucleophilic addition of the amino group on the Δ16 double bond of the steroidal substrate. It is probable that the great electron donating ability of SiMe3 makes the nitrogen of compound 2 nucleophilic enough to attack the electron deficient Δ16 bond of steroid 1.
Experimental Section
General – 1H and 13C-NMR spectra were recorded in CDCl3 on a Varian Inova 400 spectrometer at 400 MHz and 100.5 MHz, respectively (Palo Alto, CA, USA). Chemical shifts are reported in ppm relative to CHCl3 (7.26 and 77.00 ppm for 1H and 13C, respectively). FAB-MS analysis was carried out on a Finnigan MAT 95SQ spectrometer in 3-nitrobenzyl-alcohol matrix (Bremen, Germany). IR spectrum was made using a Thermo Nicolet Avatar 330 FT-IR instrument (Madison, WI, USA). The sample was prepared as a KBr pellet. Elemental analysis was performed on a Carlo Erba EA 1008 instrument (Milan, Italy).
Synthesis of dimer 3 – 1 mmol steroid 1 (356 mg) and 1.5 mmol O-(trimethylsilyl)hydroxylamine (2) (157.5 mg) was reacted in the presence of 2 mmol NaOAc (164 mg) in 10 mL refluxing methanol for 2 h. During the reaction a white precipitate was formed. After cooling the reaction mixture to room temperature, the precipitate was filtered, washed with 2 × 2 mL methanol and dried in vacuum to produce dimer 3 in 47% yield.
1H-NMR (400 MHz, CDCl3): δ = 5.35 (m, 2H, 6-H, 6’-H), 4.58 (m, 2H, 3-H, 3’-H), 3.96 (m, 2H, 16-H, 16’-H), 2.77 (d, J = 8 Hz, 2H, 17-H, 17’-H), 2.30 (m, 4H, 4-H2, 4’-H2); 2.12 (s, 6H, 21-H3, 21’-H3); 2.01 (s, 6H, CH3COO-); 1.00–2.00 (m, 31 H, ring protons, OH), 0.99 (s, 6H, 19-H3, 19’-H3), 0.63 (s, 6H, 18-H3, 18’-H3) ppm. 13C-NMR (100.5 MHz, CDCl3): δ = 208.1 (C20, C20’), 170.3 (CH3COO-C3, CH3COO-C3’), 139.3 (C5, C5’), 122.1 (C6, C6’), 73.6 (C3, C3’), 68.0 (C17, C17’), 65.8 (C16, C16’), 55.0 (C14, C14’), 49.5 (C9, C9’), 44.0 (C13, C13’), 38.4 (C12, C12’), 37.8 (C4, C4’), 36.7 (C15, C15’), 36.5 (C10, C10’), 36.4 (C1, C1’), 31.8 (C21, C21’), 31.5 (C8, C8’), 31.4 (C7, C7’), 27.5 (C2, C2’), 21.2 (CH3COO-C3, CH3COO-C3’), 20.5 (C11, C11’), 19.0 (C19, C19’), 14.1 (C18, C18’) ppm. IR (KBr): ν = 3480, 1730, 1705, 1440, 1380, 1250, 1040 cm−1. FAB-MS m/z: 746 (M+H)+. Anal. calcd. for C46H67NO7: C, 74.06; H, 9.05; N, 1.88; found C, 73.88; H, 9.09; N, 1.91.

{kind=link}
{kind=link}
{kind=link}
{kind=link}