A one-step synthesis of pyrazolone

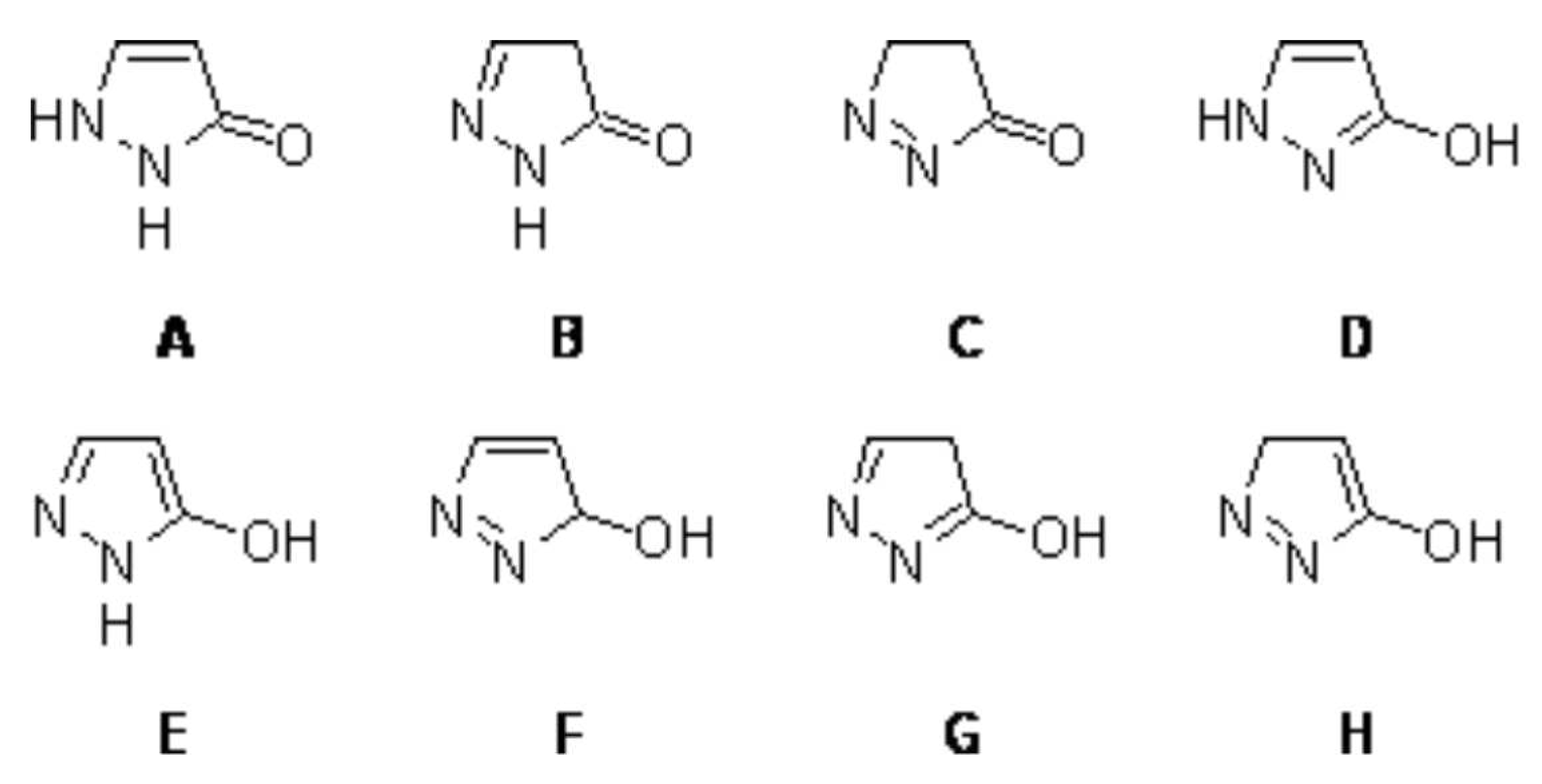{kind=link}
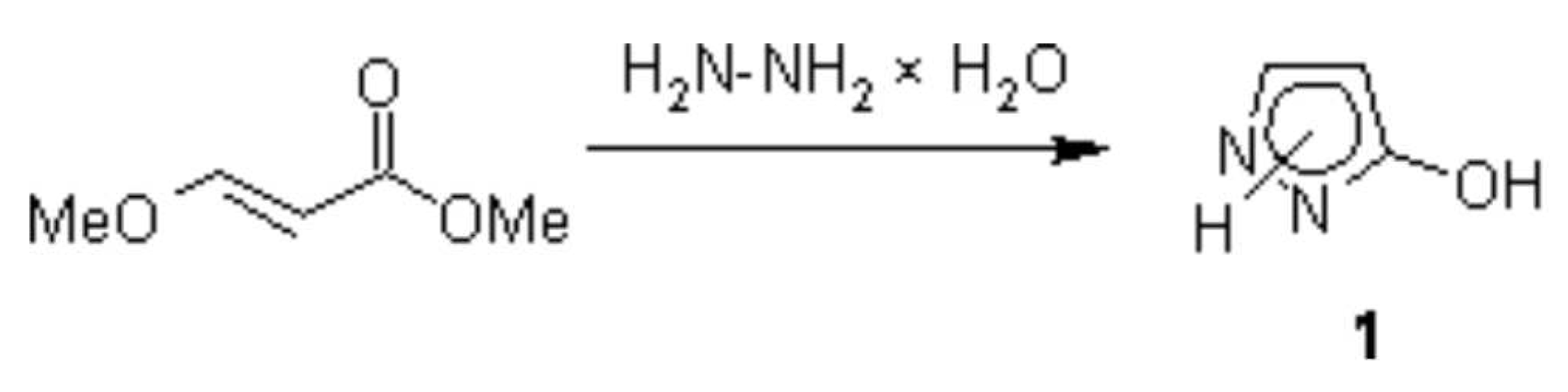{kind=link}
Abstract
:Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3References and Notes:
- Elguero, J. Comprehensive Heterocyclic Chemistry: Pyrazoles and their Benzo Derivatives; Vol. 5, Katritzky, A. R., Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; pp. 167–303. [Google Scholar]
- Stanovnik, B.; Svete, J. Product class 1: Pyrazoles. Science of Synthesis 2002, 12, 15–225. [Google Scholar] [CrossRef]
- Eller, G. A.; Holzer, W. Heterocycles 2004, 63, 2537–2555.
- Becker, W.; Eller, G. A.; Holzer, W. Synthesis 2005, 2583–2589.
- Testa, E.; Fontanella, L. Farmaco 1971, 26, 1017–35.
- Dorn, H.; Zubek, A. J. Prakt. Chem. 1971, 313, 1118–24. [CrossRef]
- Maywald, V.; Steinmetz, A.; Rack, M.; Gotz, N.; Gotz, R.; Henkelmann, J.; Becker, H. Aiscar Bayeto, PCT Int. Appl. WO 0031042 A2 2000 (Chem. Abstr., 2000, 133, 4655).
- Holzer, W.; Hallak, L. Heterocycles 2004, 63, 1311–1334, and references cited therein.
- Cizmarik, J.; Lycka, A. Pharmazie 1988, 43, 794–795. [PubMed]
- Holzer, W.; Kautsch, C.; Laggner, C.; Claramunt, R. M.; Perez-Torralba, M.; Alkorta, I.; Elguero, J. Tetrahedron 2004, 60, 6791–6805.
- Sackus, A.; Holzer, W. manuscript in preparation.
- Lingens, F.; Schneider-Bernloehr, H. Liebigs Ann. Chem. 1965, 686, 134–144.
- The spectrum was obtained on a Varian UnityPlus 300 spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C). The center of the solvent signal was used as an internal standard which was related to TMS with δ 2.49 ppm (1H NMR) and δ 39.5 ppm (13C NMR).
- The spectrum was obtained on a Bruker Avance 500 spectrometer and was referenced against neat, external nitromethane (coaxial capillary). The signals were not unequivocally assigned to the N atoms.
- The spectrum was obtained on a Shimadzu QP 1000 instrument (EI, 70eV).
- Sample Availability: Available from MDPI.
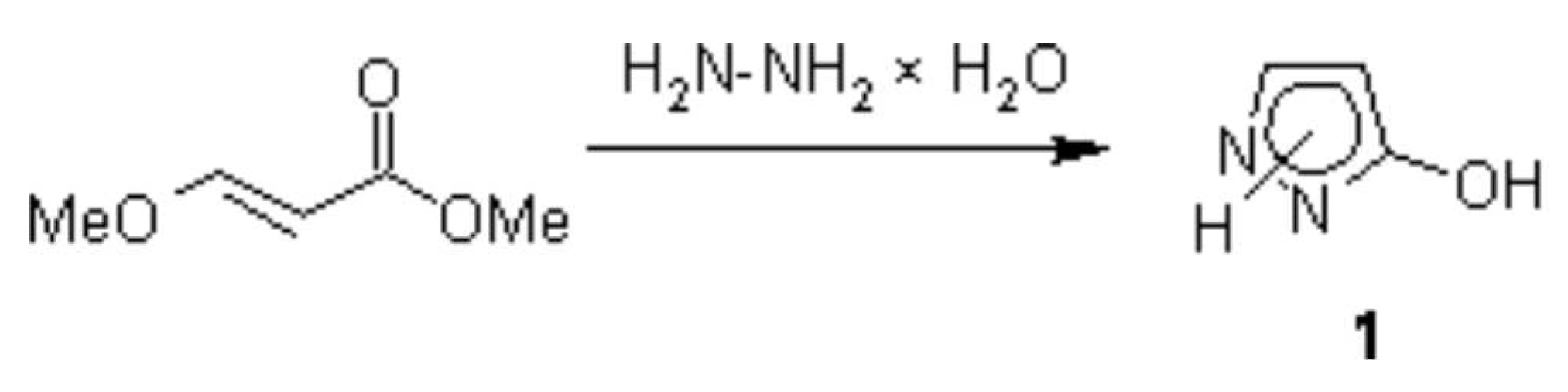
© 2006 MDPI. All rights reserved.
Share and Cite
Eller, G.A.; Holzer, W. A one-step synthesis of pyrazolone. Molbank 2006, 2006, M464. https://doi.org/10.3390/M464
Eller GA, Holzer W. A one-step synthesis of pyrazolone. Molbank. 2006; 2006(1):M464. https://doi.org/10.3390/M464
Chicago/Turabian StyleEller, Gernot A., and Wolfgang Holzer. 2006. "A one-step synthesis of pyrazolone" Molbank 2006, no. 1: M464. https://doi.org/10.3390/M464
APA StyleEller, G. A., & Holzer, W. (2006). A one-step synthesis of pyrazolone. Molbank, 2006(1), M464. https://doi.org/10.3390/M464