QSAR Study of p56lck Protein Tyrosine Kinase Inhibitory Activity of Flavonoid Derivatives Using MLR and GA-PLS

Abstract
:1. Introduction
2. Results and Discussion
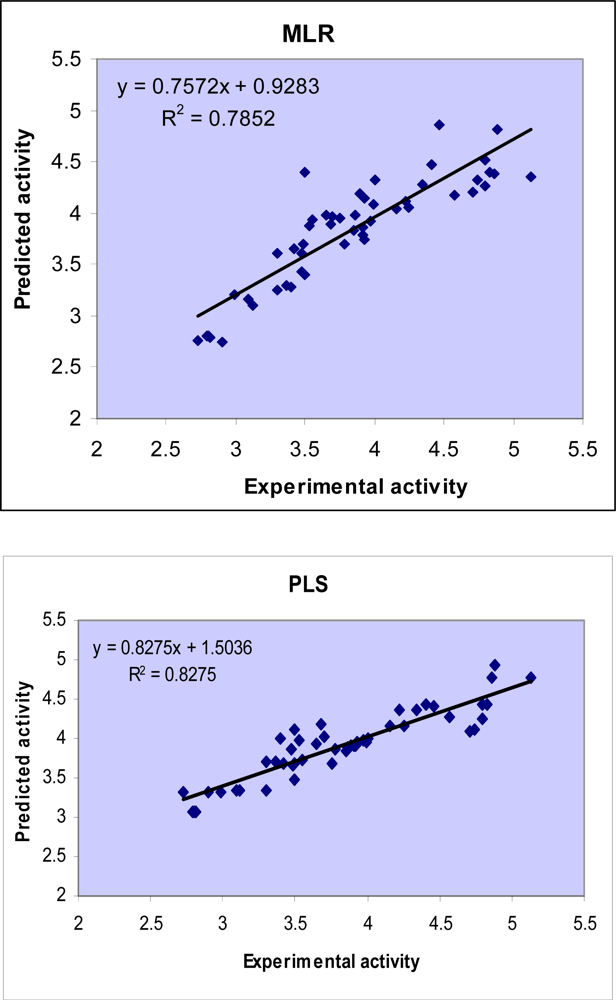
2.1. MLR analysis
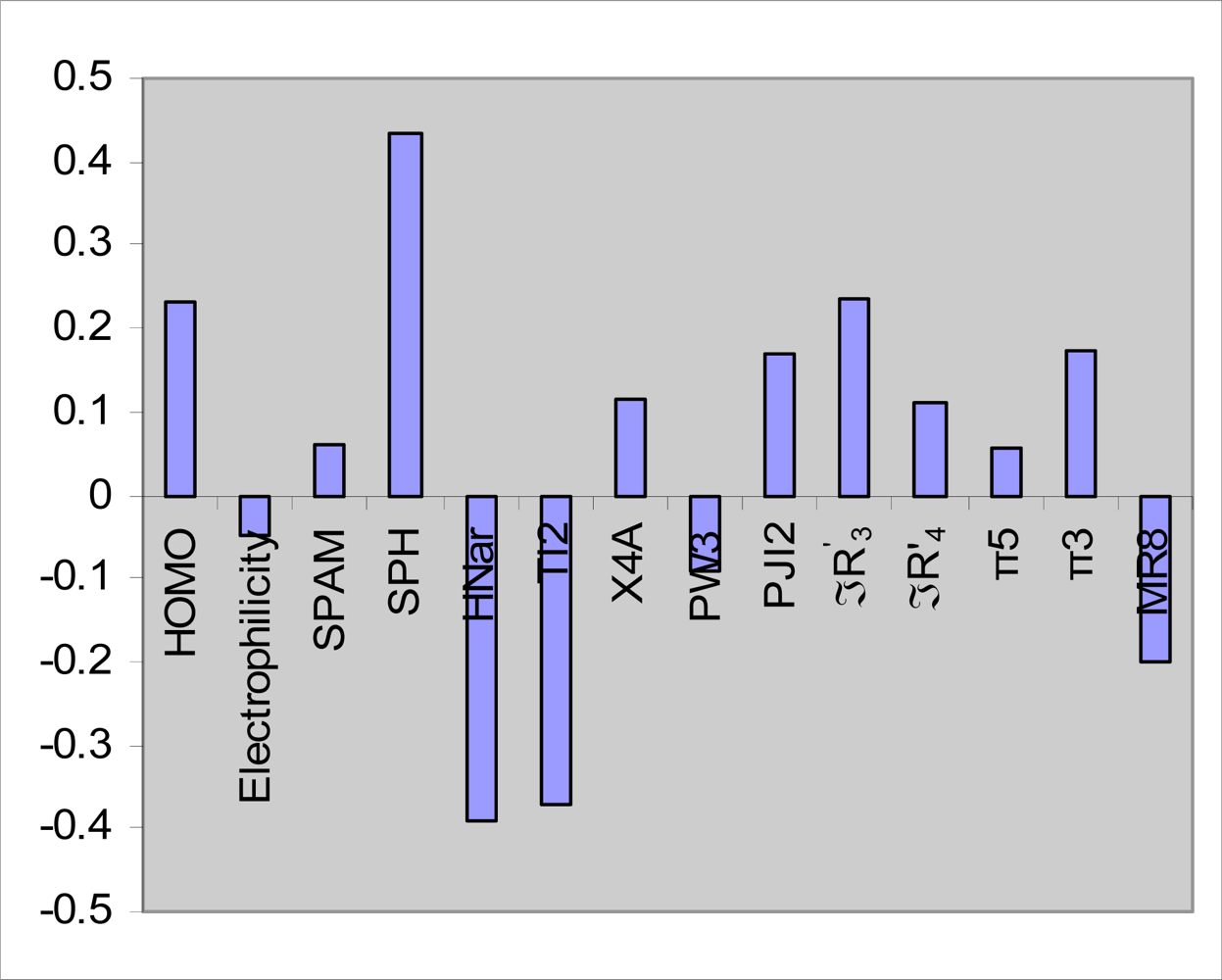
2.2. Free-Wilson analysis
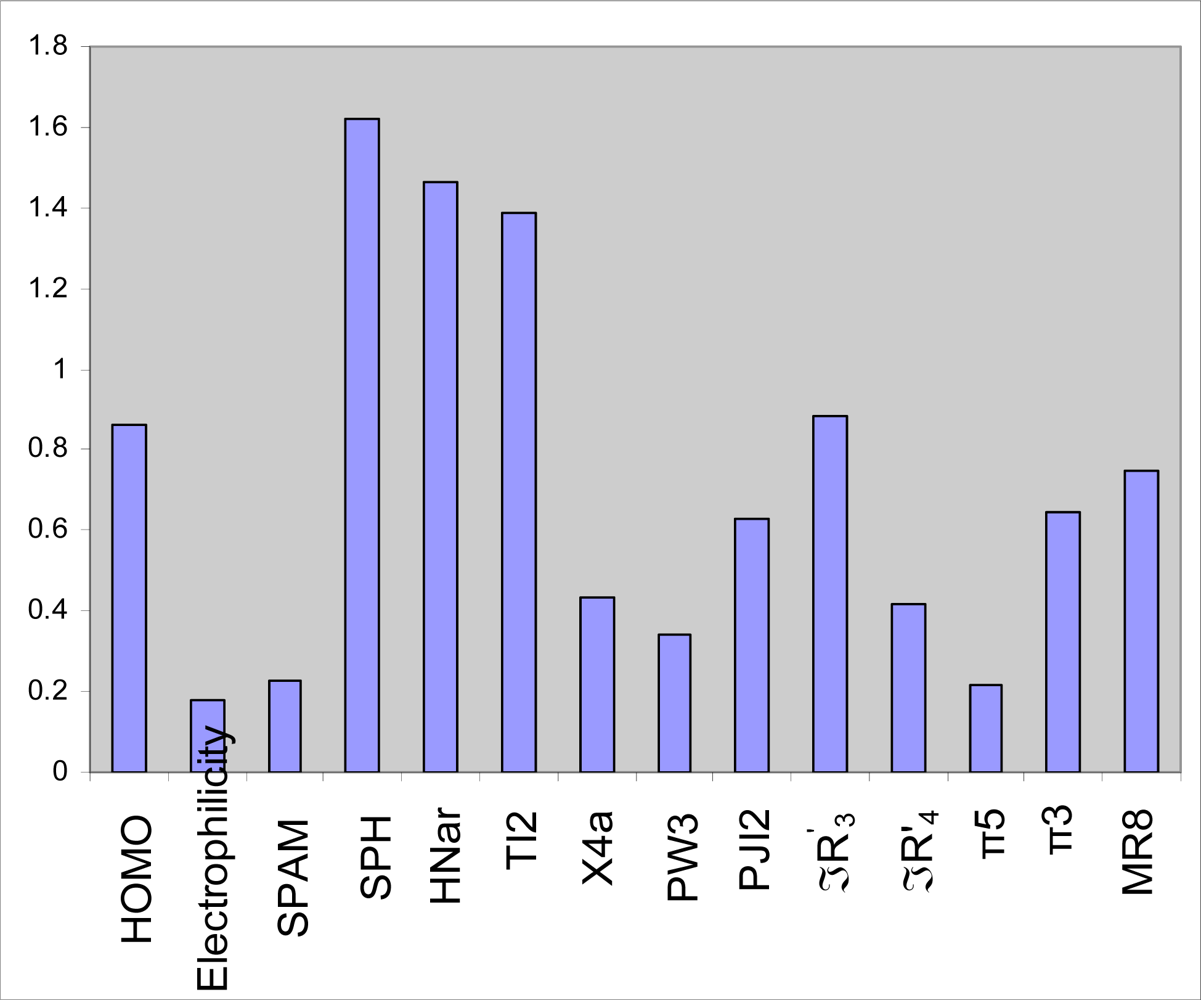
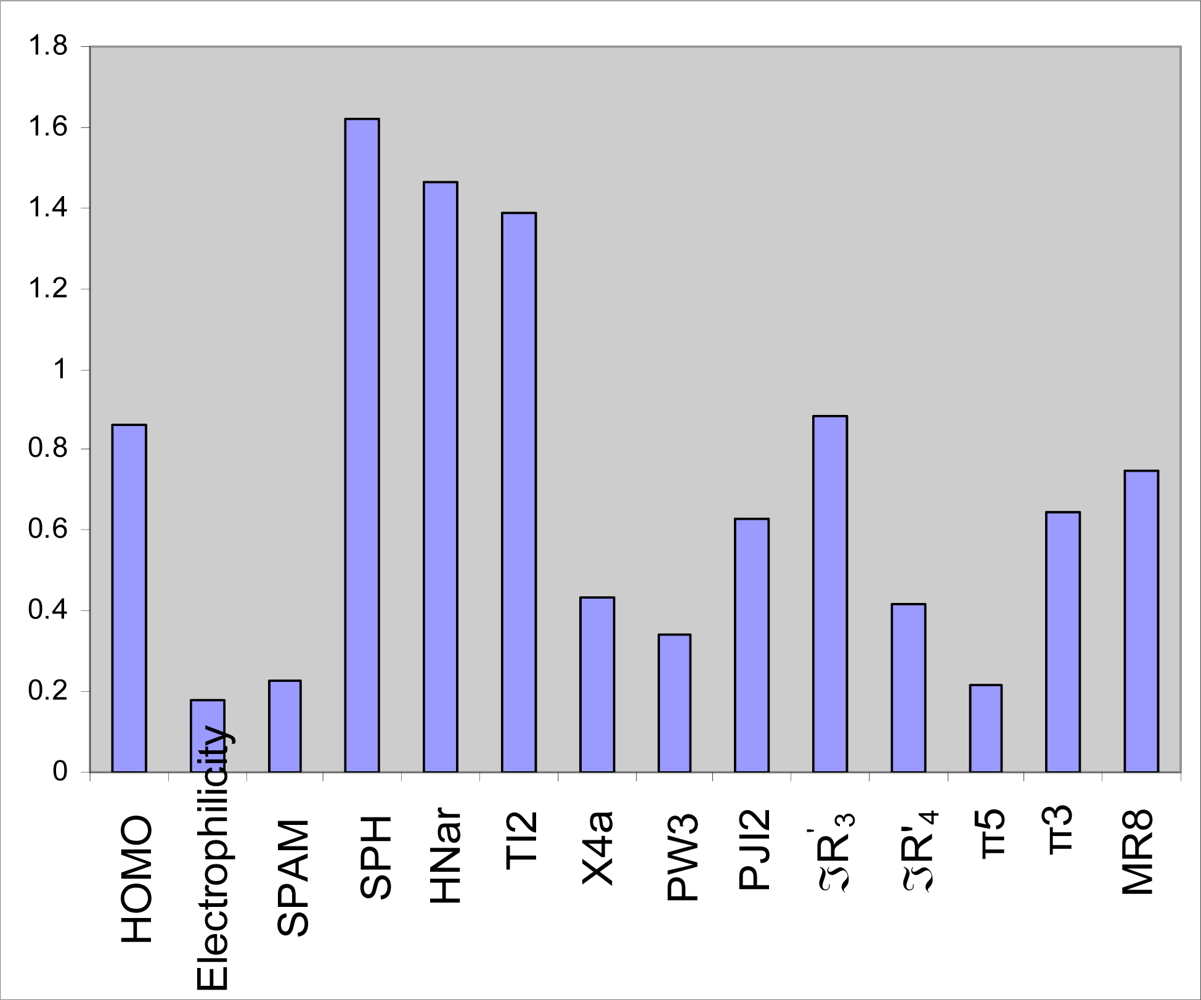
2.3. GA-PLS analysis
3. Methodology
3.1. Software
3.2. Activity data & descriptor generation
3.3. Data screening & model building
3.4. Variable importance in the projection (VIP)
3.5. Substituent electronic descriptors (SED)
4. Conclusions
Acknowledgments
References
- Hansch, C; Hoekman, D; Gao, H. Comparative QSAR: Toward a Deeper Understanding of Chemicobiological Interactions. Chem. Rev 1996, 96, 1045–1076. [Google Scholar]
- Hansch, C; Maloney, PP; Fujita, T; Muir, RM. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar]
- Hemmateenejad, B. Correlation Ranking Procedure for Factor Selection in PC-ANN Modeling and Application to ADMETox Evaluation. Chemom. Intell. Lab. Syst 2005, 75, 231–245. [Google Scholar]
- Fujita, T; Iwasa, J; Hansch, C. A New Substituent Constant, π, Derived from Partition Coefficients. J. Am. Chem. Soc 1964, 86, 5175–5180. [Google Scholar]
- Hansch, C. Quantitative Approach to Biochemical Structure-Activity Relationships. Acc. Chem. Res 1968, 2, 232–239. [Google Scholar]
- Hansch, C; Clayton, JM. Lipophilic Character and Biological Activity of Drugs II: The Parabolic Case. J. Pharm. Sci 1973, 62, 1–21. [Google Scholar]
- Agatonovic-Kustrin, S; Tucker, IG; Zecevic, M; Ziva-novic, LJ. Prediction of Drug Transfer into Human Milk from Theoretically Derived Descriptors. Anal. Chem. Acta 2000, 418, 181–195. [Google Scholar]
- Mohajeri, A; Hemmateenejad, B; Mehdipour, A; Miri, R. Modeling Calcium Channel Antagonistic Activity of Dihydropyridine Derivatives Using QTMS Indices Analyzed by GA-PLS and PC-GA-PLS. J. Mol. Graph. Model 2008, 26, 1057–1065. [Google Scholar]
- Ullrich, A; Schlessinger, J. Signal Transduction by Receptors with Tyrosine Kinase Activity. Cell 1990, 61, 203–212. [Google Scholar]
- Bishop, JM. The Molecular Genetics of Cancer. Science 1987, 235, 305–311. [Google Scholar]
- Blume-Jensen, P; Hunter, T. Oncogenic Kinase C Signalling. Nature 2001, 411, 355–365. [Google Scholar]
- Hunter, T. Signaling — 2000 and Beyond. Cell 2000, 100, 113–127. [Google Scholar]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar]
- Hanahan, D; Weinberg, RA. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Cantley, LC; Auger, KR; Carpenter, C; Duckworth, B; Graziani, A; Kapeller, R; Oltoff, S. Oncogenes and Signal Transduction. Cell 1991, 64, 281–302. [Google Scholar]
- Groundwater, PW; Solomons, KRH; Drewe, JA; Munawar, MA. Progress in Medicinal Chemistry; Ellis, GP, Luscombe, DK, Eds.; Elsevier Science B.V: Amsterdam, 1996; pp. 233–329. [Google Scholar]
- Bolen, JB; Veillette, A; Schwartz, AM; DeSeau, V; Rosen, N. Activation of pp60c-src Protein Kinase Activity in Human Colon Carcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 2251–2255. [Google Scholar]
- Slamon, DJ; Clark, GM; Wong, SG; Levin, WJ; Ullrich, A; McGuire, WL. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/neu Oncogene. Science 1987, 235, 177–182. [Google Scholar]
- Yamamoto, T; Kamata, N; Kawano, H; Shimizu, S; Kuroki, T; Toyoshima, K; Rikimaru, K; Nomura, N; Ishizaki, R; Pastan, I; Gamou, S; Shimizu, N. High Incidence of Amplification of the Epidermal Growth Factor Receptor Gene in Human Squamous Carcinoma Cell Lines. Cancer Res 1986, 46, 414–416. [Google Scholar]
- Weil, R; Veillette, A. Signal Transduction by the Lymphocyte-Specific Tyrosine Protein Kinase p56lck. Current Topics Micro. Immunol 1996, 205, 63–87. [Google Scholar]
- Anderson, SJ; Levin, SD; Perlmutter, RM. Involvement of the Protein Tyrosine Kinase p56lck in T Cell Signaling and Thymocyte Development. Adv. Immunol 1994, 56, 151–178. [Google Scholar]
- Bishop, JM. Cellular Oncogenes and Retroviruses. Annue. Rev. Biochem 1983, 52, 301–354. [Google Scholar]
- Cushman, M; Nagarathnam, D; Burg, DL; Geahlen, RL. Synthesis and Protein-Tyrosine Kinase Inhibitory Activities of Flavonoid Analogues. J. Med. Chem 1991, 34, 798–806. [Google Scholar]
- Cushman, M; Zhu, H; Geahlen, RL; Kraker, AJ. Synthesis and Biochemical Evaluation of a Series of Aminoflavones as Potential Inhibitors of Protein-Tyrosine Kinases p56lck, EGFr, and p60v-src. J. Med. Chem 1994, 37, 3353–3362. [Google Scholar]
- Bylka, W; Matlawska, I; Pilewski, NA. Natural Flavonoids as Antimicrobial Agents. JANA 2004, 7, 24–31. [Google Scholar]
- Thakur, A; Vishwakarma, S; Thakur, M. QSAR Study of Flavonoid Derivatives as p56lck Tyrosine Kinase Inhibitors. Bioorg. Med. Chem 2004, 12, 1209–1214. [Google Scholar]
- Nikolovska-Coleska, Ž; Suturkova, L; Dorevski, K; Krbavcic, A; Solmajer, T. Quantitative Structure-Activity Relationship of Flavonoid Inhibitors of p56lck Protein Tyrosine Kinase: A Classical/Quantum Chemical Approach. Quant. Struct.-Act. Relat 1998, 17, 7–13. [Google Scholar]
- Novic, M; Nikolovska-Coleska, Ž; Šolmajer, T. Quantitative Structure-Activity Relationship of Flavonoid p56lck Protein Tyrosine Kinase Inhibitors. A Neural Network Approach. J. Chem. Inf. Comput. Sci 1997, 37, 990–998. [Google Scholar]
- Oblak, M; Randic, M; Solmajer, T. Quantitative Structure-Activity Relationship of Flavonoid Analogues.3. Inhibition of p56lck Protein Tyrosine Kinase. J. Chem. Inf. Comput. Sci 2000, 40, 994–1001. [Google Scholar]
- Stefanic-Petek, A; Krbavcic, A; Solmajer, T. QSAR of Flavonoids: 4. Differential Inhibition of Aldose Reductase and p56lck Protein Tyrosine Kinase. Croatica Chemica Acta 2002, 75, 517–529. [Google Scholar]
- Meyer, M. Ab initio Study of Flavonoid. Int. J. Quantum Chem 2000, 76, 724–732. [Google Scholar]
- Deeb, O; Clare, BW. QSAR of Aromatic Substances: Protein Tyrosin Kinase Inhibitory Activity of Flavonoid Analogues. Chem. Biol. Drug Des 2007, 70, 437–449. [Google Scholar]
- Free, SMJR; Wilson, JW. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem 1964, 7, 395–399. [Google Scholar]
- Olah, M; Bologa, C; Oprea, TI. An Automated PLS Search for Biologically Relevant QSAR Descriptors. J. Comput. Aided Mol. Des 2004, 18, 437–449. [Google Scholar]
- Todeschini, R. Milano Chemometrics and QSPR Group. http://michem.disat.unimib.it/, accessed 9 September, 2008.
- Frisch, MJ; Trucks, MJ; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Zakrzewski, VG; Montgomery, JA; Stratmann, JR; Burant, JC; et al. Gaussian 98, Revision A.7. Gaussian, Inc: Pittsburgh, PA, 1998. [Google Scholar]
- Roy, K. QSAR of Adenosine Receptor Antagonists II: Exploring Physicochemical Requirements for Selective Binding of 2-arylpyrazolo [3,4-c]quinoline Derivatives with Adenosine A1 and A3 Receptor Subtypes. QSAR. Comb. Sci 2003, 22, 614–621. [Google Scholar]
- Hansch, C; Leo, A; Taft, RW. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- Bhattacharya, P; Roy, K. QSAR of Adenosine A3 Receptor Antagonist 1,2,4-triazolo[4,3-a]quinoxalin-1-one Derivatives Using Chemometric Tools. Bioorg. Med. Chem. Lett 2005, 15, 3737–3743. [Google Scholar]
- Leardi, R. Genetic Algorithms in Chemometrics and Chemistry: A Review. J. Chemometrics 2001, 15, 559–569. [Google Scholar]
- Hemmateenejad, B. Optimal QSAR Analysis of the Carcinogenic Activity of Drugs by Correlation Ranking and Genetic Algorithm-Based. J. Chemometrics 2004, 18, 475–485. [Google Scholar]
- Wang, J; Zhang, L; Yang, G; Zhan, CG. Quantitative Structure-Activity Relationship for Cyclic Imide Derivatives of Protoporphyrinogen Oxidase Inhibitors: A Study of Quantum Chemical Descriptors from Density Functional Theory. J. Chem. Inf. Comput. Sci 2004, 44, 2099–2105. [Google Scholar]
- Hemmateenejad, B; Sanchooli, M. Substituent Electronic Descriptors for Fast QSAR/QSPR. J. Chemometrics 2007, 21, 96–107. [Google Scholar]
- Smeyers, YG; Bouniam, L; Smeyers, NJ; Ezzamarty, A; Hernandez-Laguna, A; Sainz-Diaz, CI. Quantum Mechanical and QSAR Study of Some a-Arylpropionic Acids as Anti-Inflammatory Agents. Eur. J. Med. Chem 1998, 33, 103–112. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
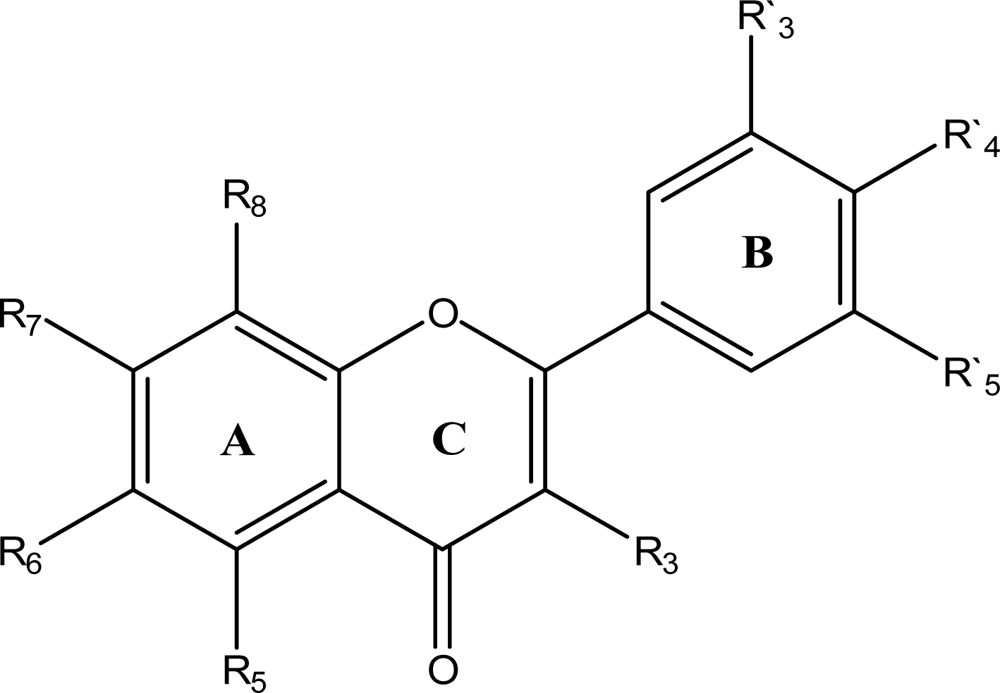{kind=link}
 Chemical structure of flavonoid derivatives.
Chemical structure of flavonoid derivatives.
| Compound | R | Experimental pIC50a | Predicted pIC50 | REP b |
|---|---|---|---|---|
| 1 | 5,7-OH,4′-NH2 | 5.13 | 4.7707 | −0.0753 |
| 2 | 3,5,7,3′,4′-OH | 4.88 | 4.9431 | 0.0128 |
| 3 | 3,7,3′,4′-OH | 4.86 | 4.7707 | −0.0187 |
| 4 | 5,7,4′-OH | 4.83 | 4.4356 | −0.0889 |
| 5 | 5,4′-OH | 4.80 | 4.2603 | −0.1267 |
| 6 | 6,3′-OH | 4.80 | 4.4242 | −0.0849 |
| 7 | 6-OH,5,7,4′-NH2 | 4.74 | 4.1061 | −0.1544 |
| 8 | 5,7-OH | 4.71 | 4.0895 | −0.1518 |
| 9 | 4′-OH,3′,5′-OCH3 | 4.57 | 4.2687 | −0.0706 |
| 10 | 5,7,3′,4′-OH | 4.46 | 4.4172 | −0.0097 |
| 11 | 7,3′-OH | 4.41 | 4.4358 | 0.0058 |
| 12 | 6-OH,5,7,3′-NH2 | 4.34 | 4.3681 | 0.0064 |
| 13 | 6-OMe,8,3′-NH2 | 4.25 | 4.1649 | −0.0204 |
| 14 | 6-OH,3′,4′,5′-OCH3 | 4.22 | 4.3591 | 0.0319 |
| 15 | 3,5,7,4′-OH,3′,5′-OCH3 | 4.16 | 4.1649 | 0.0012 |
| 16 | 3,5,7,3′,5′-OH | 4.00 | 3.9947 | −0.0013 |
| 17 | 6,4′-NH2 | 3.99 | 3.9613 | −0.0072 |
| 18 | 6,8,4′-NH2 | 3.97 | 3.9764 | 0.0016 |
| 19 | 6-OH,8,4′-NH2 | 3.93 | 3.9446 | 0.0037 |
| 20 | 6,4′-OH | 3.93 | 3.9247 | −0.0013 |
| 21 | 7,8,4′-OH,3′,5′-OCH3 | 3.92 | 3.8990 | −0.0054 |
| 22 | 8,4′-NH2 | 3.91 | 3.8994 | −0.0027 |
| 23 | 6,4′-OH,3′,5′-OCH3 | 3.89 | 3.9133 | 0.0060 |
| 24 | 7-OH,4′-NH2 | 3.86 | 3.8815 | 0.0056 |
| 25 | 7-OH,6,4′-NH2 | 3.85 | 3.8296 | −0.0053 |
| 26 | 7,4′-OH | 3.78 | 3.8621 | 0.0213 |
| 27 | 7,8,3′OH | 3.75 | 3.6903 | −0.0162 |
| 28 | 6,3′-NH2 | 3.70 | 4.0228 | 0.0803 |
| 29 | 4′-NH2 | 3.68 | 4.1850 | 0.1207 |
| 30 | 5-OH,6,4′-NH2 | 3.65 | 3.9325 | 0.0718 |
| 31 | 3,5,7-OH | 3.53 | 3.9794 | 0.1129 |
| 32 | 5,4′-OH,7-OCH3 | 3.55 | 3.7315 | 0.0487 |
| 33 | 5,3′-OH | 3.50 | 4.1209 | 0.1507 |
| 34 | 7,8-OH | 3.50 | 3.4873 | −0.0036 |
| 35 | 5-OH,8,4′-NH2 | 3.49 | 3.6705 | 0.0492 |
| 36 | 7-OH,8,4′-NH2 | 3.48 | 3.6694 | 0.0516 |
| 37 | 7-OH | 3.47 | 3.8567 | 0.1003 |
| 38 | 6-OCH3,8,4′-NH2 | 3.43 | 3.6709 | 0.0683 |
| 39 | 7,8-OH,3′,4′,5′-OCH3 | 3.40 | 4.0058 | 0.1512 |
| 40 | 3-COOCH3,4′-OH | 3.36 | 3.7081 | 0.0939 |
| 41 | 4′-OH | 3.30 | 3.7081 | 0.1101 |
| 42 | 7-OH,6,3′-NH2 | 3.30 | 3.3419 | 0.0125 |
| 43 | 7-OH,6,8,4′-NH2 | 3.12 | 3.3419 | 0.0664 |
| 44 | 3-COOCH3,4′-NH2 | 3.09 | 3.3419 | 0.0754 |
| 45 | 3-COOH,7-OCH3,4′-OH | 2.99 | 3.3262 | 0.1011 |
| 46 | 7,4′-OH,3′,5′-OCH3 | 2.90 | 3.3262 | 0.1281 |
| 47 | 7-OH,6,8,4′-NO2 | 2.81 | 3.0674 | 0.0839 |
| 48 | 3-COOH,4′-OH | 2.80 | 3.0674 | 0.0872 |
| 49 | 5-OCH3,8,4′-NH2 | 2.79 | 3.0674 | 0.0904 |
| 50 | 7-OH,8,4′-NO2 | 2.73 | 3.3262 | 0.1793 |
| Descriptor type | Molecular Description |
|---|---|
| Constitutional | Molecular weight, no. of atoms, no. of non-H atoms, no. of bonds, no. of heteroatoms, no. of multiple bonds (nBM), no. of aromatic bonds, no. of functional groups (hydroxyl, amine, aldehyde, carbonyl, nitro, nitroso, etc.), no. of rings, no. of circuits, no of H-bond donors, no of H-bond acceptors, no. of Nitrogen atoms (nN), chemical composition, sum of Kier-Hall electrotopological states (Ss), mean atomic polarizability (Mp), number of rotable bonds (RBN), mean atomic Sanderson electronegativity (Me), etc.
|
| Topological | Molecular size index, molecular connectivity indices (X1A, X4A, X2v, X1Av, X2Av, X3Av, X4Av), information content index (IC), Kier Shape indices, total walk count, path/walk-Randic shape indices (PW3, PW4, Zagreb indices, Schultz indices, Balaban J index (such as MSD) Wiener indices, topological charge indices, Sum of topological distances between F..F (T(F..F)), Ratio of multiple path count to path counts (PCR), Mean information content vertex degree magnitude (IVDM), Eigenvalue sum of Z weighted distance matrix (SEigZ), reciprocal hyper-detour index (Rww), Eigenvalue coefficient sum from adjacency matrix (VEA1), radial centric information index, 2D petijean shape index (PJI2), etc.
|
| Geometrical | 3D petijean shape index (PJI3), Gravitational index, Balaban index, Wiener index, etc.
|
| Quantum | Highest occupied Molecular Orbital Energy (HOMO) , Lowest Unoccupied Molecular Orbital Energy (LUMO), Most positive charge (MPC), Least negative charge (LNC), Sum of squares of charges (SSC), Sum of square of positive charges (SSPC), Sum of square of negative charges (SSNC), Sum of positive charges (SUMPC), Sum of negative charges (SUMNC), Sum of absolute of charges (SAC), Total dipole moment (DMt), Molecular dipole moment at X-direction (DMX), Molecular dipole moment at Y-direction (DMY), Molecular dipole moment at Z-direction (DMZ), Electronegativity (χ= −0.5 (HOMO-LUMO)), Electrophilicity (ω= χ2/2 η) ,Hardness (η = 0.5 (HOMO+LUMO)), Softness (S=1/η).
|
| Functional group | Number of total tertiary carbons (nCt), Number of H-bond acceptor atoms (nHAcc), number of total hydroxyl groups (nOH), number of unsubstituted aromatic C(nCaH), number of ethers (aromatic) (nRORPh), etc.
|
| Chemical | LogP (Octanol-water partition coefficient), Hydration Energy (HE), Polarizability (Pol), Molar refractivity (MR), Molecular volume (V), Molecular surface area (SA).
|
| Substituent electronic descriptors | RMSQ (Root mean square error of charges), SPQ ( Sum of positive charges), SNQ ( Sum of negative charges), RMSDM (Root mean square of dipole moments at any Cartesian coordinate direction), TDM (Total dipole moment), FRMS (Root mean square force that any atom in constituent molecule see right before the optimization), FMAX (Maximum force on molecule), HOMO (Highest occupied molecular orbital), LUMO (Lowest unoccupied molecular orbital), HD (Hardness), SOF (Softness), EPH (Electrophilicity), EN (Electronegativity). |
| No. | Descriptor source | MLR Equations | N | R2 | SE | RMSCV | Q2 | F |
|---|---|---|---|---|---|---|---|---|
| E1 | Chemical | pIC50 = 4.893 (± 0.735) − 0.056 (± 0.017) HE −0.007 (± 0.003) Mass | 50 | 0.40 | 0.55 | 0.58 | 0.32 | 13.82 |
| E2 | Quantum | pIC50 = 6.362 (± 0.565) − 6.805 (± 1.505) MPC | 50 | 0.43 | 0.53 | 0.54 | 0.38 | 17.44 |
| E3 | Constitutional | pIC50 = 3.139 (± 1.250) − 0.438 (± 0.100) nBM − 0.506 (± 0.205) AMW − 0.584 (± 0.266) nAB | 50 | 0.49 | 0.49 | 0.51 | 0.42 | 19.65 |
| E4 | Topological | pIC50 = 17.242 (± 0.605) − 3.374 (± 0.545) IVDM − 53.95 (± 12.355) X1Av + 2.349 (± 0.696) ICR +24.874 (±9.569) PW4 + 73.575 (±33.719) X4A | 50 | 0.72 | 0.38 | 0.48 | 0.58 | 30.13 |
| E5 | Geometrical | pIC50 = −15.093 (± 3.339) + 19.450 (± 3.406) SPH − 0.010 (± 0.002) G(N...O) | 50 | 0.60 | 0.43 | 0.47 | 0.49 | 17.23 |
| E6 | Functional group | pIC50 = 3.672 (± 0.123) − 0.414 (± 0.130) nNO2 −1.098 (± 0.369) nOHt + 0.160 (± 0.058) nOH | 50 | 0.53 | 0.45 | 0.50 | 0.45 | 12.67 |
| E7 | Hansch | pIC50 = 4.219 (± 0.289) − 0.615 (± 0.202) π5 + 1.462 (± 0.555) ℑR′3 − 1.379 (± 0.490) ℑR8 −0.249 (± 0.111) L3 | 50 | 0.53 | 0.45 | 0.50 | 0.45 | 12.67 |
| E8 | SED | pIC50 = −0.708 (± 1.228) − 9.570 (± 2.500) HOMOA3 + 1.092 (±0.308) SNQ8 | 50 | 0.82 | 0.32 | 0.30 | 0.61 | 51.43 |
| E9 | Molecular descriptor | pIC50 = −19.763 (± 4.304) − 4.785 (± 1.275) MPC + 25.113 (± 4.142) SPH + 0.849 (± 0.264) SNQ8 − 0.357 (± 0.136) L3 | 50 | 0.83 | 0.31 | 0.28 | 0.62 | 52.43 |
| HE | Mass | MPC | nBM | AMW | nAB | ASP | G(N...O) | X1AV | ICR | PW4 | X4A | IVDM | nNO2 | nOHt | nOH | ℑR′3 | L3 | ℑR8 | π5 | pIC50 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HE | 1 | −0.234 | 0.192 | 0.124 | −0.327 | 0.236 | −0.006 | 0.00 | 0.651 | 0.075 | −0.012 | 0.316 | 0.065 | 0.069 | 0.047 | −0.745 | −0.394 | 0.067 | −0.005 | 0.485 | −0.347 |
| Mass | 1 | 0.531 | 0.580 | 0.512 | 0.136 | −0.269 | 0.328 | −0.655 | 0.416 | 0.541 | −0.631 | 0.816 | 0.554 | 0.099 | 0.211 | 0.326 | 0.196 | 0.487 | 0.040 | −0.268 | |
| MPC | 1 | 0.953 | 0.715 | 0.366 | −0.233 | 0.623 | −0.539 | 0.304 | 0.050 | −0.329 | 0.904 | 0.876 | 0.259 | −0.227 | −0.286 | 0.289 | 0.595 | 0.156 | −0.547 | ||
| nBM | 1 | 0.778 | 0.165 | −0.094 | 0.725 | −0.624 | 0.390 | 0.016 | −0.325 | 0.937 | 0.972 | 0.114 | −0.196 | −0.211 | 0.125 | 0.687 | 0.193 | −0.498 | |||
| AMW | 1 | 0.050 | −0.200 | 0.356 | 0.897 | 0.037 | 0.116 | −0.206 | 0.718 | 0.775 | 0.116 | 0.434 | 0.136 | 0.125 | 0.620 | 0.065 | −0.191 | ||||
| nAB | 1 | −0.684 | −0.127 | 0.069 | −0.192 | 0.257 | −0.397 | 0.235 | −0.073 | 0.692 | −0.086 | −0.198 | 0.930 | −0.108 | 0.185 | −0.364 | |||||
| ASP | 1 | 0.294 | 0.155 | 0.538 | 0.532 | 0.388 | −0.221 | 0.069 | 0.369 | −0.273 | −0.201 | −0.768 | −0.039 | −0.098 | 0.269 | ||||||
| G(N...O) | 1 | −0.379 | 0.578 | 0.299 | 0.348 | 0.618 | 0.763 | −0.138 | −0.478 | −0.437 | −0.182 | 0.508 | 0.034 | −0.329 | |||||||
| X1AV | 1 | −0.130 | −0.171 | 0.413 | −0.651 | −0.647 | −0.052 | −0.572 | −0.270 | −0.056 | −0.542 | 0.229 | 0.058 | ||||||||
| ICR | 1 | −0.212 | −0.277 | 0.442 | 0.441 | −0.104 | −0.410 | −0.161 | −0.278 | 0.153 | 0.168 | −0.080 | |||||||||
| PW4 | 1 | −0.157 | 0.261 | −0.045 | 0.158 | 0.336 | 0.413 | 0.356 | 0.029 | −0.249 | 0.002 | ||||||||||
| X4A | 1 | −0.489 | −0.233 | −0.252 | −0.046 | −0.025 | −0.466 | −0.261 | −0.157 | 0.347 | |||||||||||
| IVDM | 1 | 0.891 | 0.155 | −0.100 | −0.030 | 0.218 | 0.663 | 0.192 | −0.494 | ||||||||||||
| nNO2 | 1 | −0.050 | −0.177 | −0.166 | −0.097 | 0.720 | 0.151 | −0.416 | |||||||||||||
| nOHt | 1 | 0.061 | −0.137 | 0.513 | −0.075 | 0.128 | −0.306 | ||||||||||||||
| nOH | 1 | 0.621 | 0.104 | −0.004 | −0.375 | 0.370 | |||||||||||||||
| ℑR′3 | 1 | −0.070 | 0.008 | −0.014 | 0.315 | ||||||||||||||||
| L3 | 1 | −0.143 | 0.085 | −0.259 | |||||||||||||||||
| ℑR8 | 1 | 0.224 | −0.367 | ||||||||||||||||||
| π5 | 1 | −0.451 | |||||||||||||||||||
| pIC50 | 1 |
© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fassihi, A.; Sabet, R. QSAR Study of p56lck Protein Tyrosine Kinase Inhibitory Activity of Flavonoid Derivatives Using MLR and GA-PLS. Int. J. Mol. Sci. 2008, 9, 1876-1892. https://doi.org/10.3390/ijms9091876
Fassihi A, Sabet R. QSAR Study of p56lck Protein Tyrosine Kinase Inhibitory Activity of Flavonoid Derivatives Using MLR and GA-PLS. International Journal of Molecular Sciences. 2008; 9(9):1876-1892. https://doi.org/10.3390/ijms9091876
Chicago/Turabian StyleFassihi, Afshin, and Razieh Sabet. 2008. "QSAR Study of p56lck Protein Tyrosine Kinase Inhibitory Activity of Flavonoid Derivatives Using MLR and GA-PLS" International Journal of Molecular Sciences 9, no. 9: 1876-1892. https://doi.org/10.3390/ijms9091876
APA StyleFassihi, A., & Sabet, R. (2008). QSAR Study of p56lck Protein Tyrosine Kinase Inhibitory Activity of Flavonoid Derivatives Using MLR and GA-PLS. International Journal of Molecular Sciences, 9(9), 1876-1892. https://doi.org/10.3390/ijms9091876