Introduction
Long-range electrostatic and van der Waals interactions have for a long time been in the center of attention in empirical methods of computational chemistry, mainly within molecular mechanics and dynamics. Particularly, electrostatic interactions belong to permanent pitfalls of computational chemistry [
1,
2,
3,
4]. There are principally two reasons for that: (i) The electrostatic and van der Waals interactions represent the most expensive part of calculations, and the evaluation of them is so time consuming that it has been necessary to use approximations; (ii) The results of the calculations are sometimes very sensitive on the approximation used.
Until recently, the most frequently used method to handle electrostatic and van der Waals interactions was to ignore all interactions between atoms whose internuclear distance is longer than a certain cutoff value. Such an approach is usually called the Cutoff Method (CM) [
5,
6,
7]. There are several possible choices concerning how the cutoff can be used. We have chosen the most straightforward definition as a distance border limit.
Since the calculation of van der Waals interactions is more expensive than that of electrostatic interactions, the CM approach can be slightly modified by using two cutoffs, one for van der Waals and the other for electrostatics [
8]. These two cutoffs may be set up in such a way, that the final accuracy of results is the same for both components. In other words, unnecessarily more precise (and expensive) calculations of one of these two types of interactions are avoided.
During last few years, Ewald mesh method and other mesh methods [
9,
10,
11,
12,
13] for example, Particle Mesh Ewald (PME) [
12,
13,
14] method, have been developed and implemented into commonly used software packages, like AMBER [
15,
16] and NWChem [
17]. These methods are able to handle electrostatic interactions efficiently. Some software packages even employ different algorithms to calculate van der Waals interactions in CM and PME approaches (for example, AMBER). PME becomes commonly used method especially for molecular dynamics simulations of nucleic acids in explicit solvent, but not only there [
7,
18,
19,
20]. Several papers have been published showing that, from the geometry point of view, PME can successfully be utilized [
21,
22,
23,
24,
25,
26]. Nevertheless, there are also papers in the literature showing that PME has some weak points, for example, some artifacts can be generated using PME [
27,
28,
29,
30,
31,
32].
As mentioned above, effects of PME method on geometrical (conformational) features of the system have several times been subjected to studies and reported in the literature. However, we have not found any paper where PME and CM energies would be subjected to such a comparison. Therefore, our effort in this work is to compare efficiency of using two cutoff values in calculations and, especially, to elucidate the speed of convergence of CM and PME energy to its final value.
Several systems have been selected for the study, representing various classes of important biomolecules.
Methods
Altogether, a total of 19 representative molecules were considered in our various testing calculations, for one of them we considered three different conformations. Those include seven peptides and proteins, eleven oligonucleotides, and three conformations of a nucleotide-sugar. More detailed structural information is shown in
Table 1.
Unless stated otherwise, all calculations were performed by using the SANDER and ANAL of AMBER 5.0 suite of programs. The initial coordinates were taken from structural databases and/or our previous calculations. First, all molecules were minimized using the distance dependent dielectric constant and the following parameters: scnb=2.0, scee=2.0, dx0=0.01, dxm=0.5, dele=0.001, drms=0.3 (remaining parameters were assigned by default values). Then, energies were calculated for various cutoff values using both the PME and cutoff methods.
To examine the dependence of the calculated energy on the used cutoff value, the energy calculations were performed using different cutoff values; the used cutoff value was increased with a step of 1 Å until all nonbonding interactions were included (denoted by the final cutoff below). For convenience, the energy calculated at the final cutoff is called the final energy.
Table 1.
General overview of studied molecules. For more details see
Table 4 and
Table 7 in Results section.
Table 1.
General overview of studied molecules. For more details see Table 4 and Table 7 in Results section.
| | | Molecule type | |
|---|
| | Peptide or protein | Oligonucleotide | Nucleotide-sugar |
|---|
| Number of molecules | 7 | 11 | 3 |
| Minimum number of atoms | 77 | 96 | 80 |
| Maximum number of atoms | 4909 | 843 | 80 |
| Number of explicitly solvated molecules | 1 | 3 | 1 |
For the energy calculation using PME, a rectangular box was created of such a size, that the calculated molecule fits the box with positive tolerance of 3 Å. We then started with a cutoff of 1 Å and subsequently increased it by 1 Å step until the value of B/2 was reached (B is the length of the shortest edge of the rectangular box). This approach was used for both explicitly and implicitly solvated systems. We have started with such a small value of cutoff since our goal was to obtain a complete image.
The calculations with explicit solvent were performed with the ANAL program that allows for separation of solute-solute, solute-solvent, and solvent-solvent interactions. The coordinates of the atoms used in these calculations were taken from equilibrated molecular dynamics (MD) snapshots.
Results
Complexity of electrostatics and van der Waals energy calculation
In this section we only discuss the complexity of the interaction energy calculations within a distance which is shorter than the cutoff. This part of calculations is the same for both cutoff and PME method. We can roughly estimate the complexity of the energy calculations by the following theoretical considerations.
Electrostatic energy is calculated using Coulomb’s law – eq. (1),
i.e. one multiplication and one division.
This is calculated for n-times and (n-1)-times for the first and the second summation, respectively. For the overall time complexity T
el(n) we can write:
where cmult and cdiv are complexities of multiplication and division, respectively. We consider rij as a constant since it is necessary for calculation both electrostatic and van der Waals energy.
The simplest approximation of van der Waals energy is Lennard-Jones potential – eq. (3):
It is clear, that we can not calculate term
using less than 4 multiplications, 2 divisions, and 1 substraction. For the overall time complexity T
vdw(n) we then have:
where csub is complexity of subtraction.
For current high performance computers division is more time-consuming than multiplication, that is slightly slower than addition and subtraction (or it is of equal speed). It is then clear from equations (2) and (4) that calculation of Evdw is more time-consuming than that of Eel in all cases.
We have written a program in C language to measure and print the ratio of time complexities for the calculations of E
vdw and E
el (see
Table 2).
Table 2.
Ratio of computational complexities of van der Waals (Tvdw(n)) and electrostatic (Tel(n)) energies calculation.
Table 2.
Ratio of computational complexities of van der Waals (Tvdw(n)) and electrostatic (Tel(n)) energies calculation.
| Platform | Operating system | Compiler | Tvdw(n) / Tel(n) |
|---|
| PC, Intel Pentium II | Windows 95 | Borland C++ 3.0 | 3.703 |
| PC, Intel Pentium II | Linux 6.1 | Cc | 3.525 |
| Sun | Solaris | Cc | 3.435 |
| Silicon Graphics | Irix 6.4 | Cc | 3.671 |
The results listed in
Table 2 agree well with the above theoretical considerations. The ratio of time complexities depends on the used hardware, but we can conclude that the calculation of E
vdw is roughly about 3 times slower than that of the corresponding E
el.
Comparison in time complexity between the cutoff and PME methods
The time complexities of the cutoff and PME energy calculation methods are shown in
Table 3.
The data in
Table 3 reveal the following remarkable trends: (i) the PME method is in all cases slower than the cutoff method; (ii) for large molecules the PME is about 30% slower.
Table 3.
Comparison of computational complexity of cutoff and PME method. Calculations were executed on a Sillicon Graphics workstation with the operating system Irix 6.1.
Table 3.
Comparison of computational complexity of cutoff and PME method. Calculations were executed on a Sillicon Graphics workstation with the operating system Irix 6.1.
| Peptides and proteins: |
| Denotation of molecule | Number of atoms | Time of calculation [s] |
| cutoff method | PME method |
| P1 | 77 | 0.28 | 1.1 |
| P2 | 328 | 0.44 | 1.06 |
| P3 | 439 | 0.59 | 1.49 |
| P4 | 648 | 0.91 | 1.88 |
| P5 | 1457 | 1.97 | 3.68 |
| P6 | 4872 | 5.46 | 9.02 |
| P7 | 4909 | 8.09 | 11.31 |
| Oligonucleotides: |
| Denotation of molecule | Number of atoms | Time of calculation [s] |
| cutoff method | PME method |
| O1 | 98 | 0.29 | 0.74 |
| O2 | 100 | 0.32 | 0.80 |
| O3 | 228 | 0.47 | 1.14 |
| O4 | 228 | 0.47 | 1.11 |
| O5 | 228 | 0.47 | 1.08 |
| O6 | 516 | 1.88 | 2.19 |
| O7 | 516 | 1.86 | 2.28 |
| O8 | 728 | 2.05 | 2.31 |
| O9 | 784 | 3.29 | 4.44 |
| O10 | 784 | 3.06 | 4.09 |
| O11 | 868 | 3.36 | 4.52 |
| Nucleotide-sugars: |
| Denotation of molecule | Number of atoms | Time of calculation [s] |
| Cutoff method | PME method |
| N1 | 80 | 0.29 | 3.43 |
| N2 | 80 | 0.29 | 1.54 |
| N3 | 80 | 0.34 | 2.56 |
Peptides and proteins
Seven molecules belonging to this class were studied (see
Table 4).
Table 4.
Studied peptides and proteins.
Table 4.
Studied peptides and proteins.
| Denotation of molecule | Number of amino acids | Number of atoms | Name of molecule |
|---|
| P1 | 5 | 77 | met-enkephalin |
| P2 | 21 | 328 | endothelin-1 |
| P3 | 32 | 439 | alpha-conotoxin |
| P4 | 46 | 648 | crambin |
| P5 | 99 | 1457 | plastocyanin |
| P6 | 312 | 4872 | haloalkandehalogenase DhlA GJ10 |
| P7 | 299 | 4909 | cyclin-dependent kinase Cdk2 |
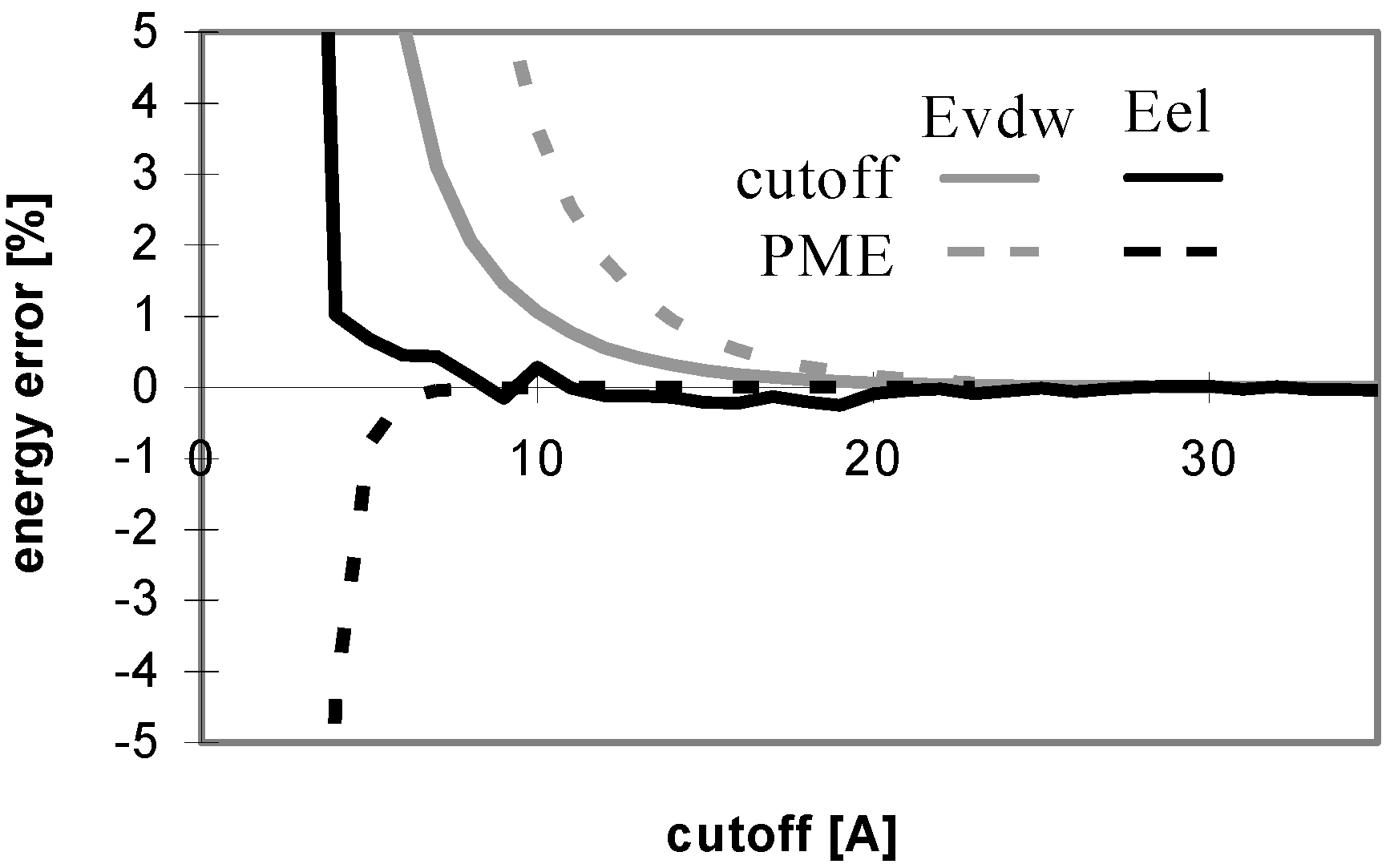
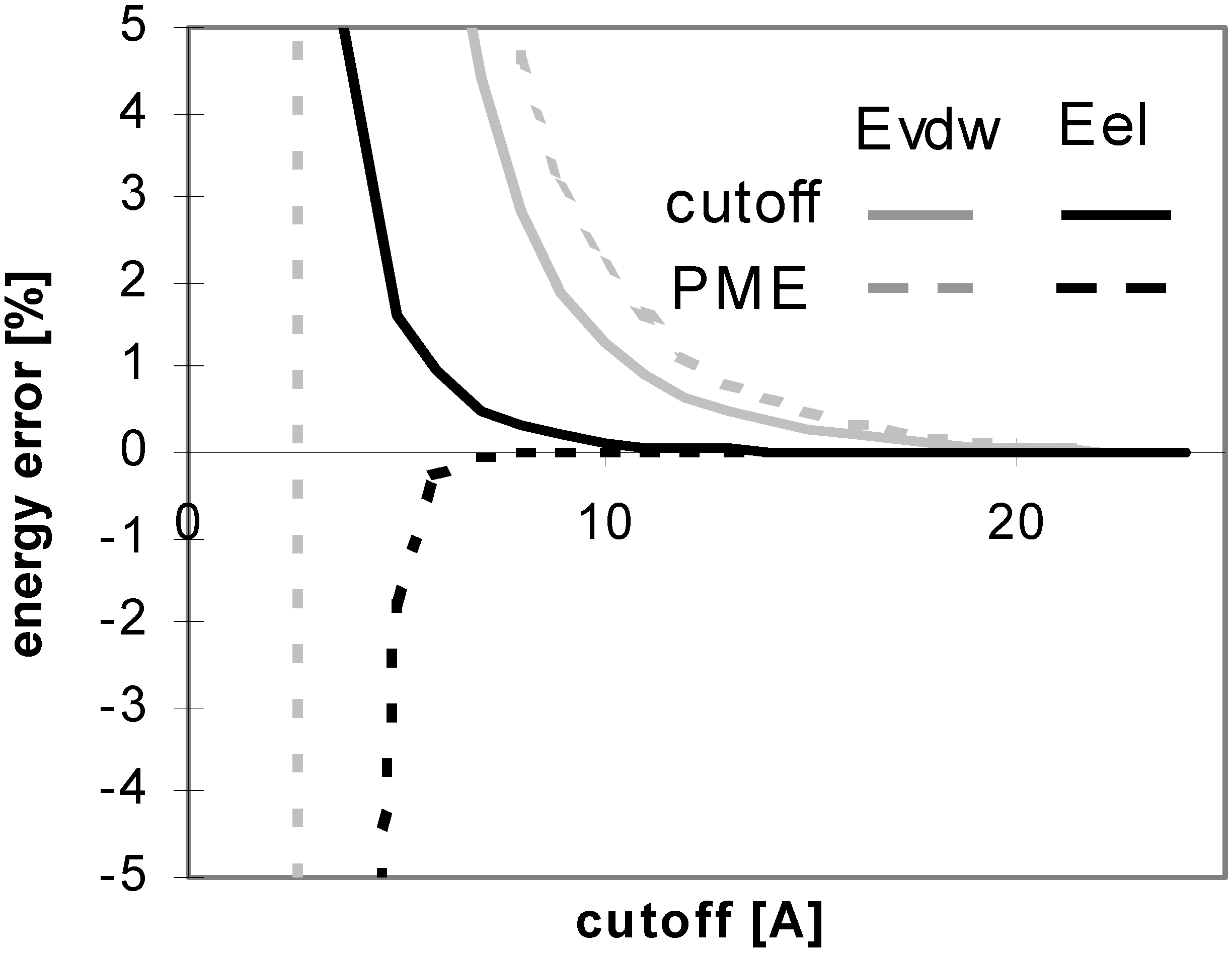
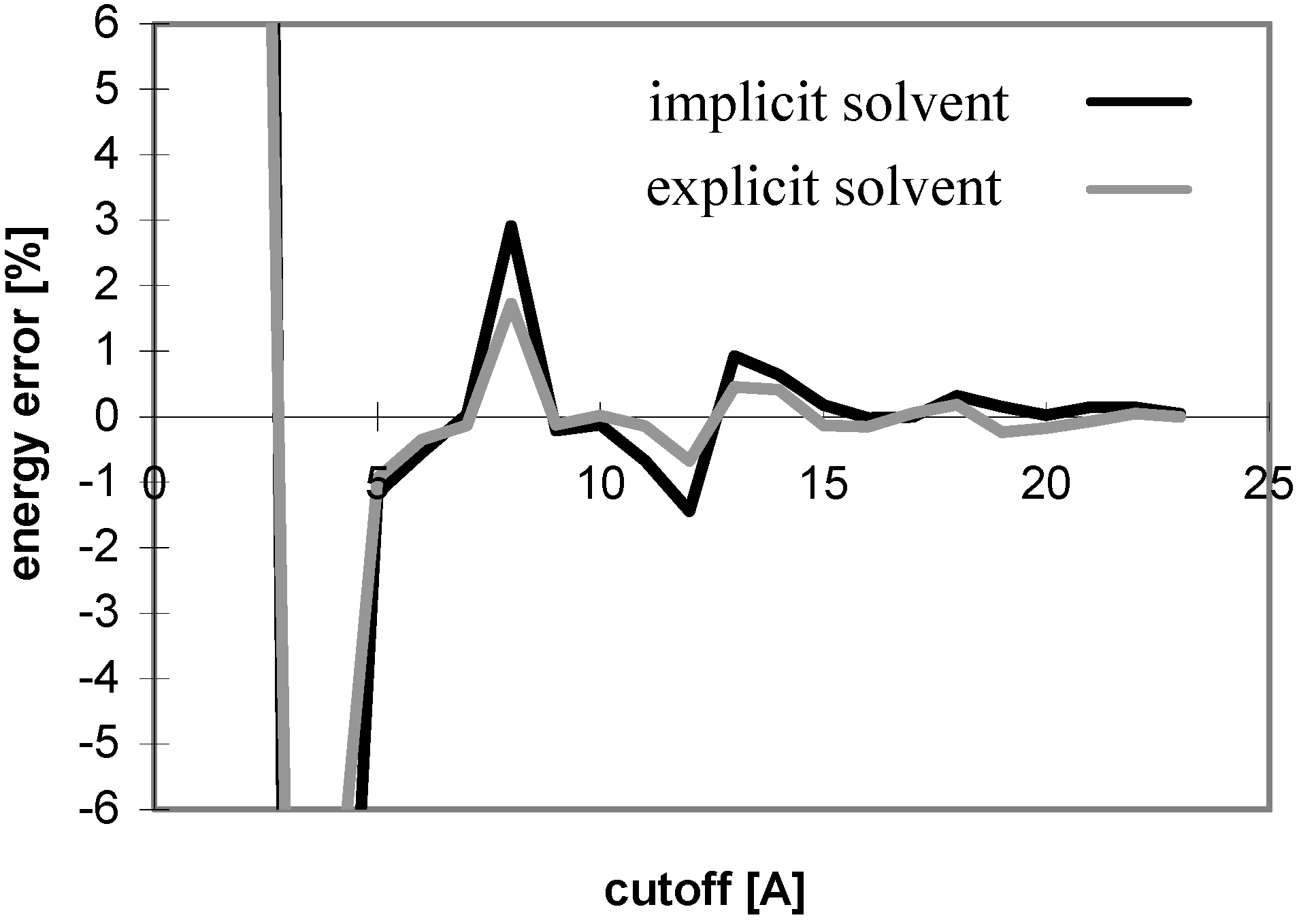
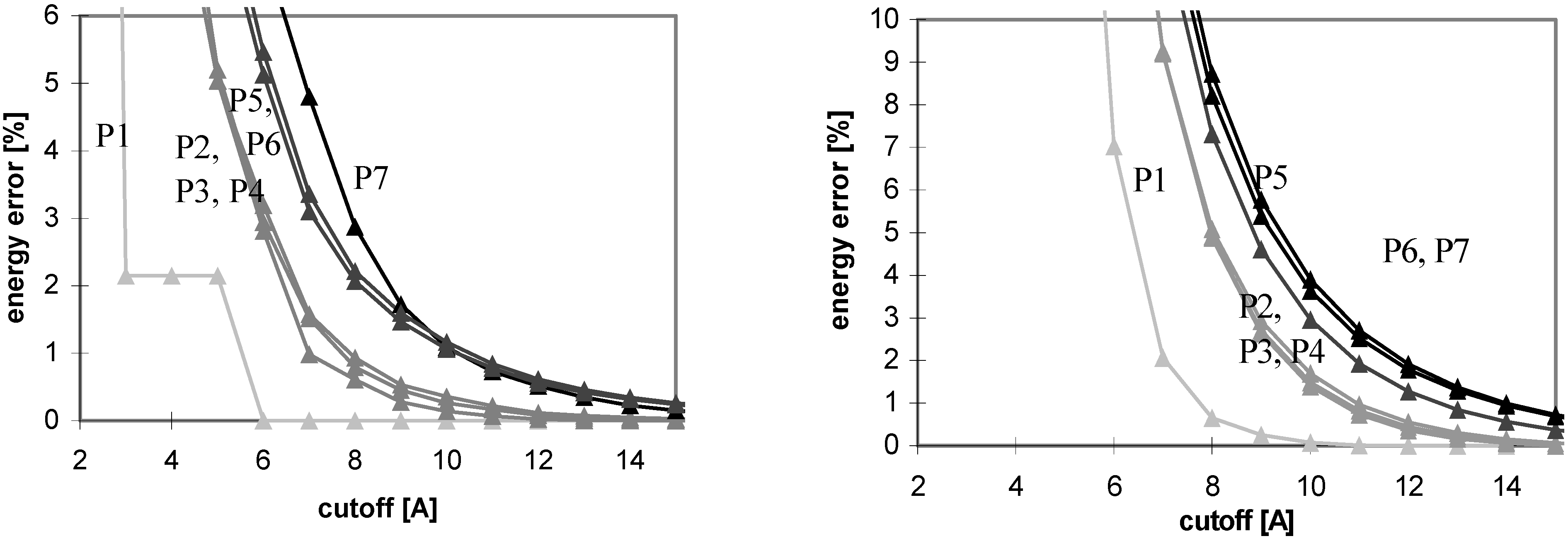
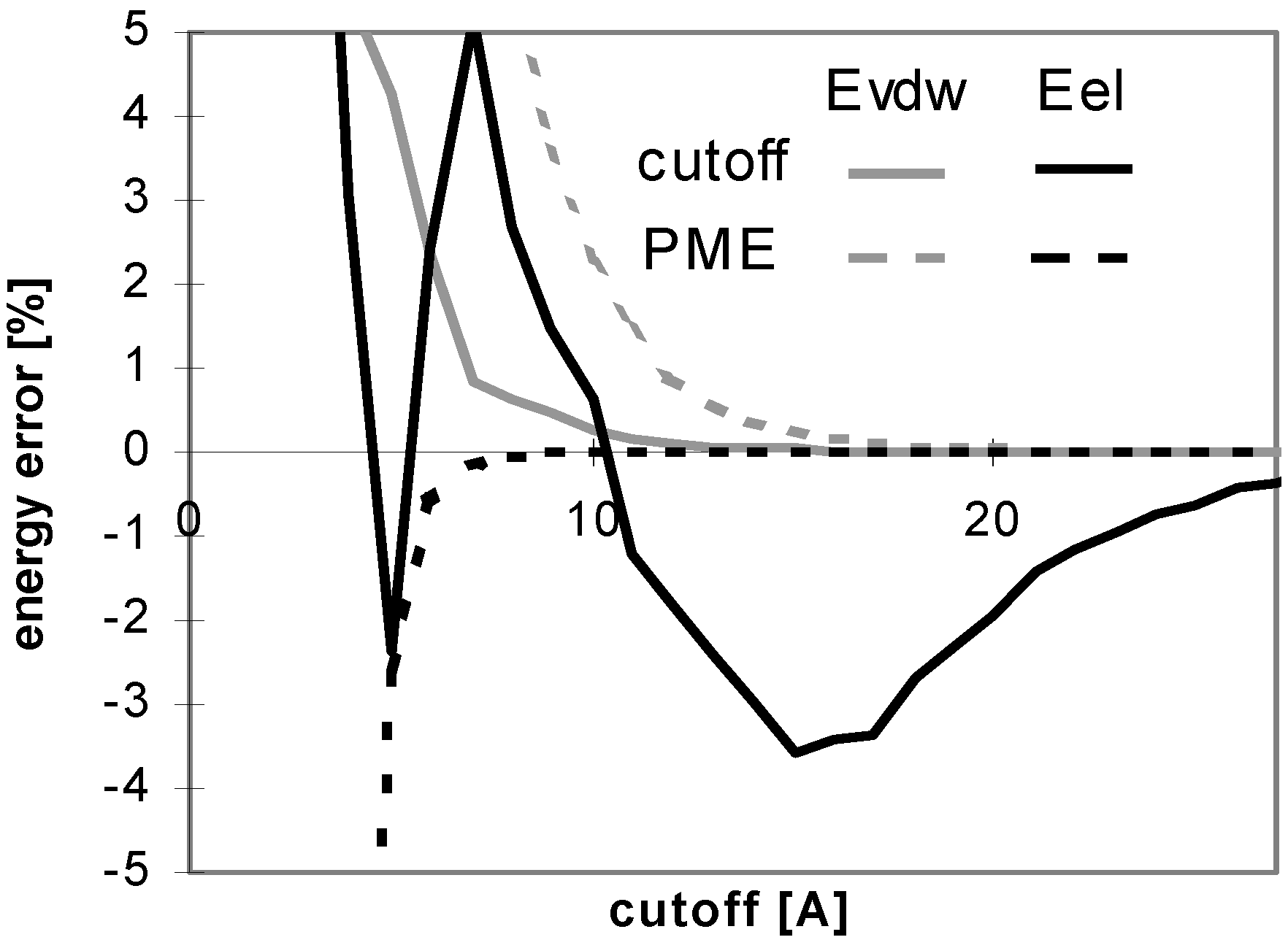
The results of energy calculations for the protein P7 are depicted in
Fig 1, which describes convergence of the energies to their final values. The results calculated for all remaining peptides and proteins are similar to what depicted in
Fig 1. The only difference is that the speed of convergence of E
vdw (for both the cutoff and PME methods) depends on the size of molecule, as illustrated in
Fig 2.
Figure 1.
Dependency of energy error on cutoff (for protein P7). Energy error (EE) is calculated using the equation: EE = 100.(E – Ef)/Ef, where E is the energy value calculated for the cutoff and Ef is the final energy (see method section).
Figure 1.
Dependency of energy error on cutoff (for protein P7). Energy error (EE) is calculated using the equation: EE = 100.(E – Ef)/Ef, where E is the energy value calculated for the cutoff and Ef is the final energy (see method section).
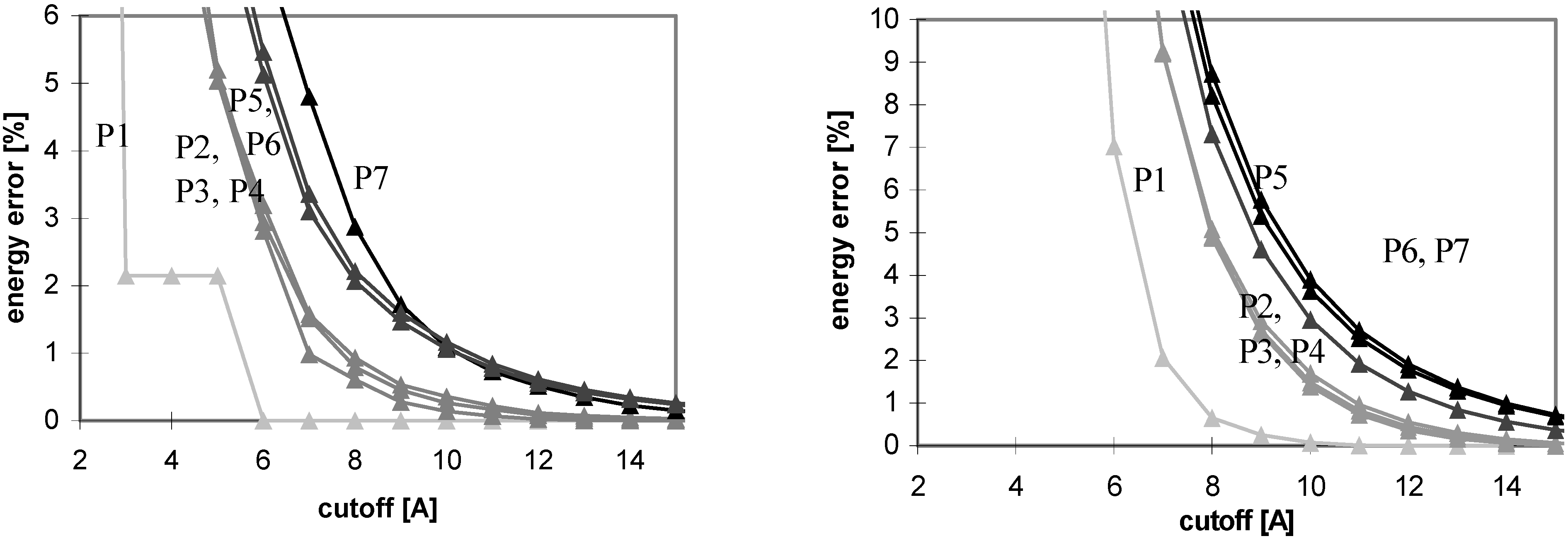
Figure 2.
Dependency of Evdw error on cutoff (for both cutoff (left) and PME (right) methods for all studied peptides and proteins, the darker color of the curve, the larger the molecule is). For description of energy error see
Figure 1.
Figure 2.
Dependency of Evdw error on cutoff (for both cutoff (left) and PME (right) methods for all studied peptides and proteins, the darker color of the curve, the larger the molecule is). For description of energy error see
Figure 1.
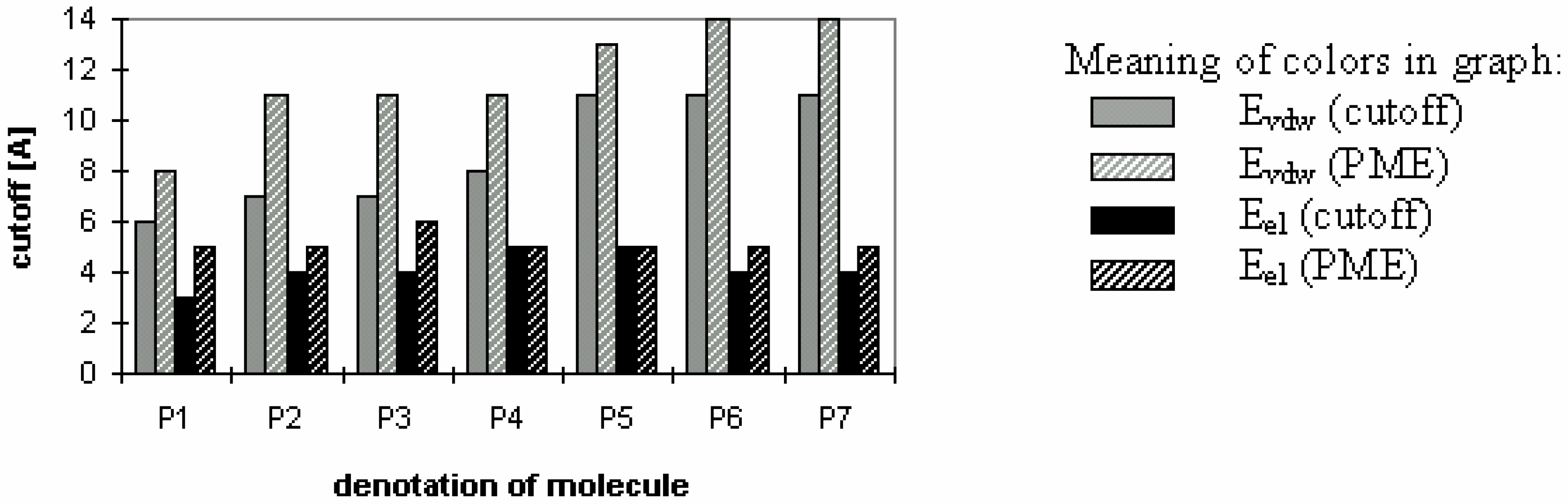
Figure 3.
Smallest values of cutoff for which energy error is smaller then 1% (for peptides and proteins).
Figure 3.
Smallest values of cutoff for which energy error is smaller then 1% (for peptides and proteins).
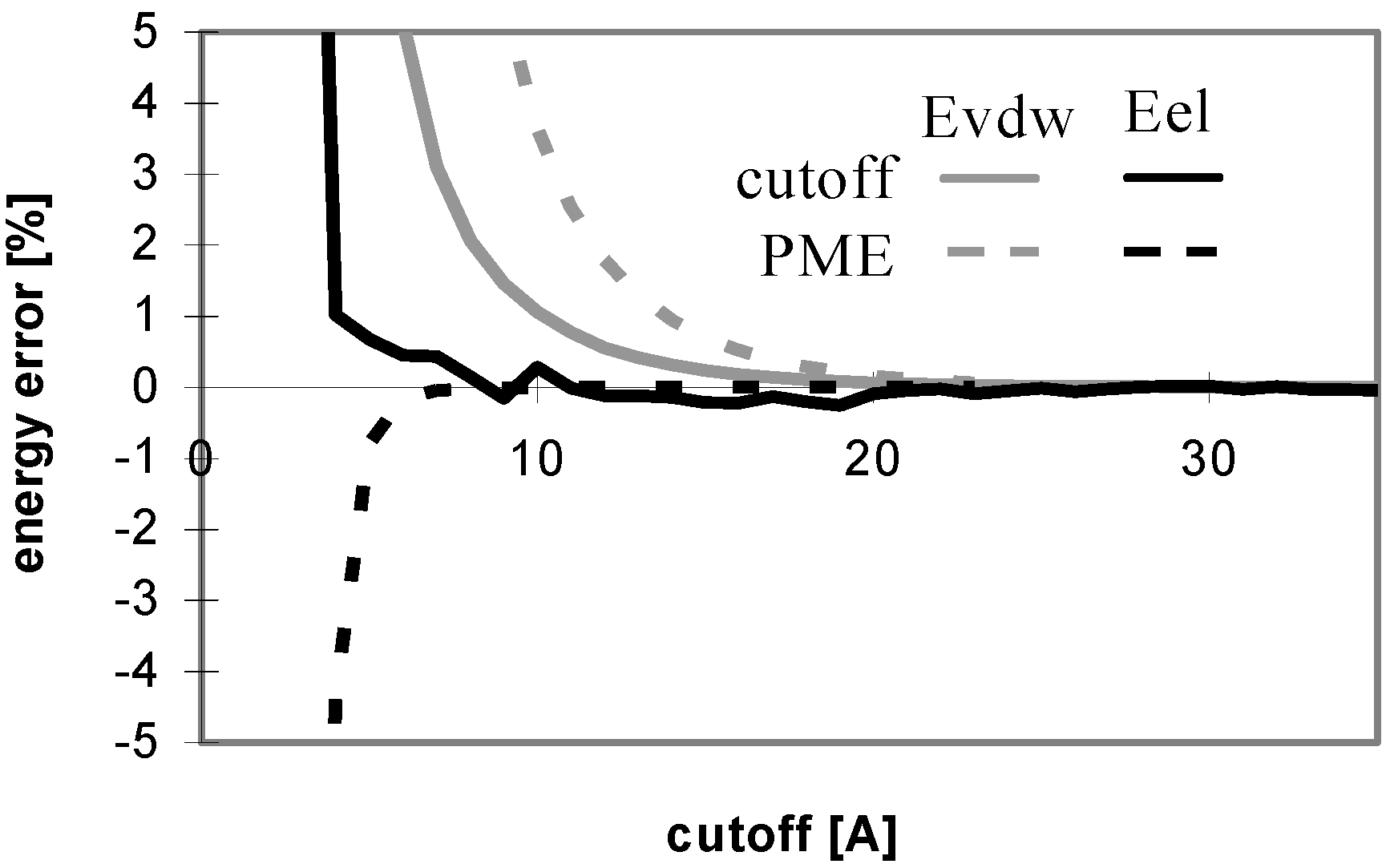
The convergence of Evdw is slower for larger molecules, i.e., they require to use a larger cutoff value. It applies for both the cutoff and PME methods. However, for PME the convergence is considerably slower. The convergence of electrostatic energy is almost independent of the size of the molecule for the considered peptides and proteins (data not shown). In terms of the energy convergence, the cutoff method seems to be better than PME for peptides and proteins.
Fig 3 shows which minimum cutoff value must be used to have the energy error below 1% for each case.
Fig 1 and
Fig 3 demonstrate that the convergence of E
vdw is markedly slower than that of E
el for both the cutoff and PME methods. A larger cutoff value is necessary for E
vdw to reach the same accuracy than for E
el. The larger the studied molecule, the greater the difference between the cutoff values required for E
vdw and E
el reaching the same accuracy of 1%. It means that the computations using different cutoff values for E
vdw and E
el will be more efficient in this case. A quite small cutoff value could be used to calculate E
el. However, this will not lead to a dramatic speed-up of the calculation since E
el is relatively inexpensive to calculate. See
Table 5 for a brief summary of these results.
Table 5.
Summary of results for peptides and proteins.
Table 5.
Summary of results for peptides and proteins.
| | Speed of Evdw convergence | Speed of Eel convergence | Evdw / Eel Convergence |
|---|
| Cutoff method | slow | fast | Eel significantly faster |
| PME method | slow | fast | Eel significantly faster |
| Cutoff / PME methods | Cutoff methods lightly faster | Cutoff method is slightly faster | Cutoff method is slightly better |
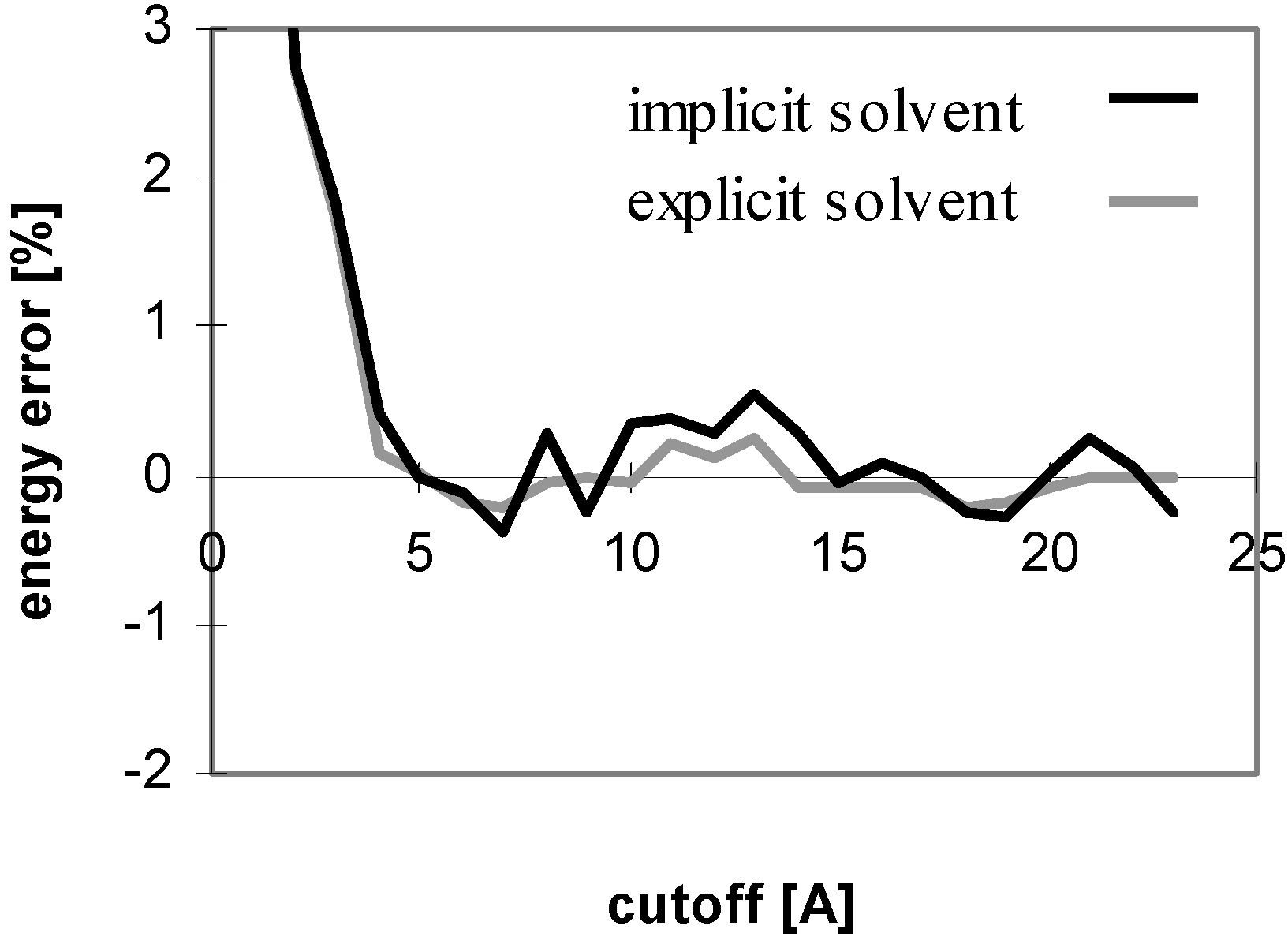
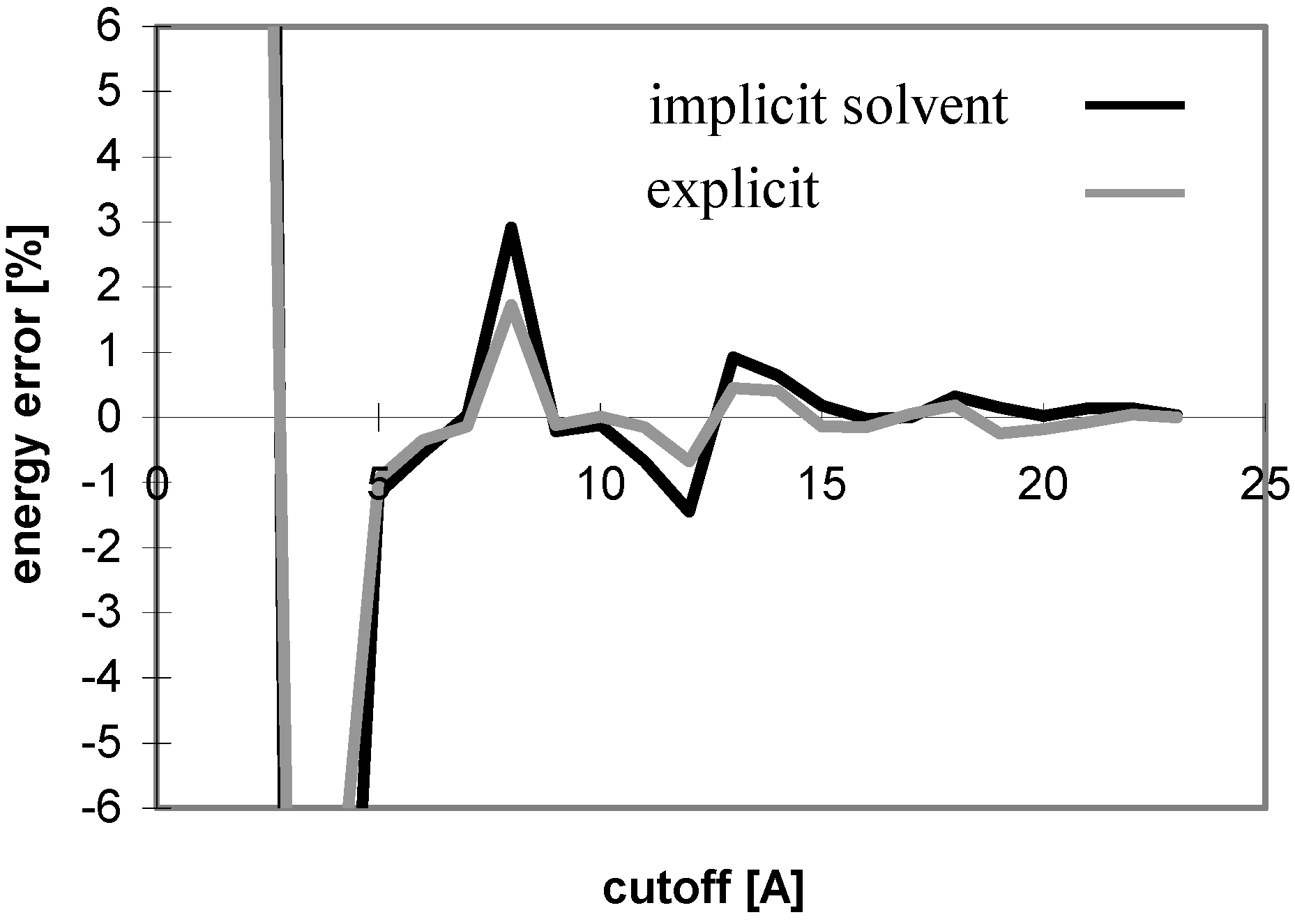
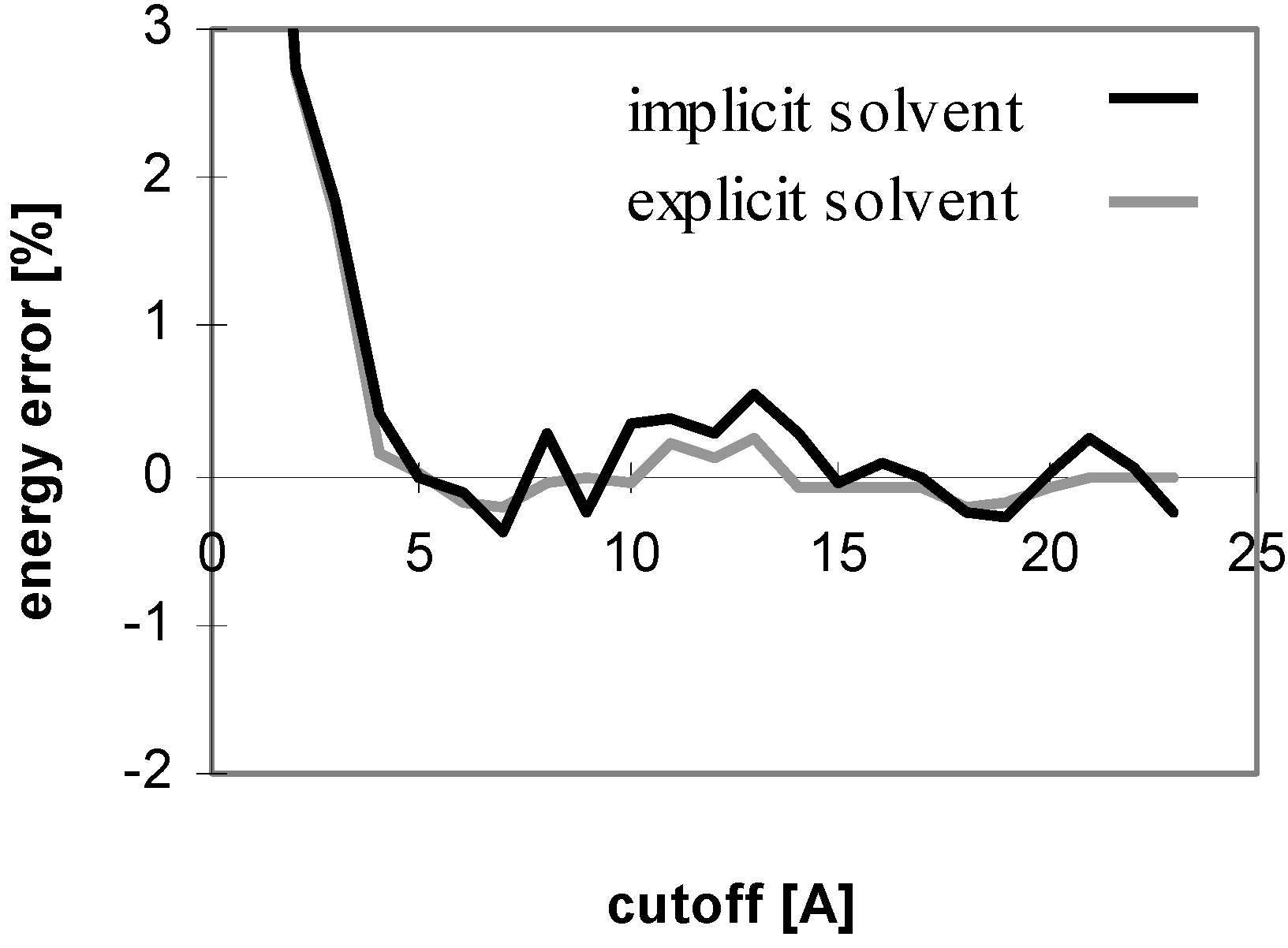
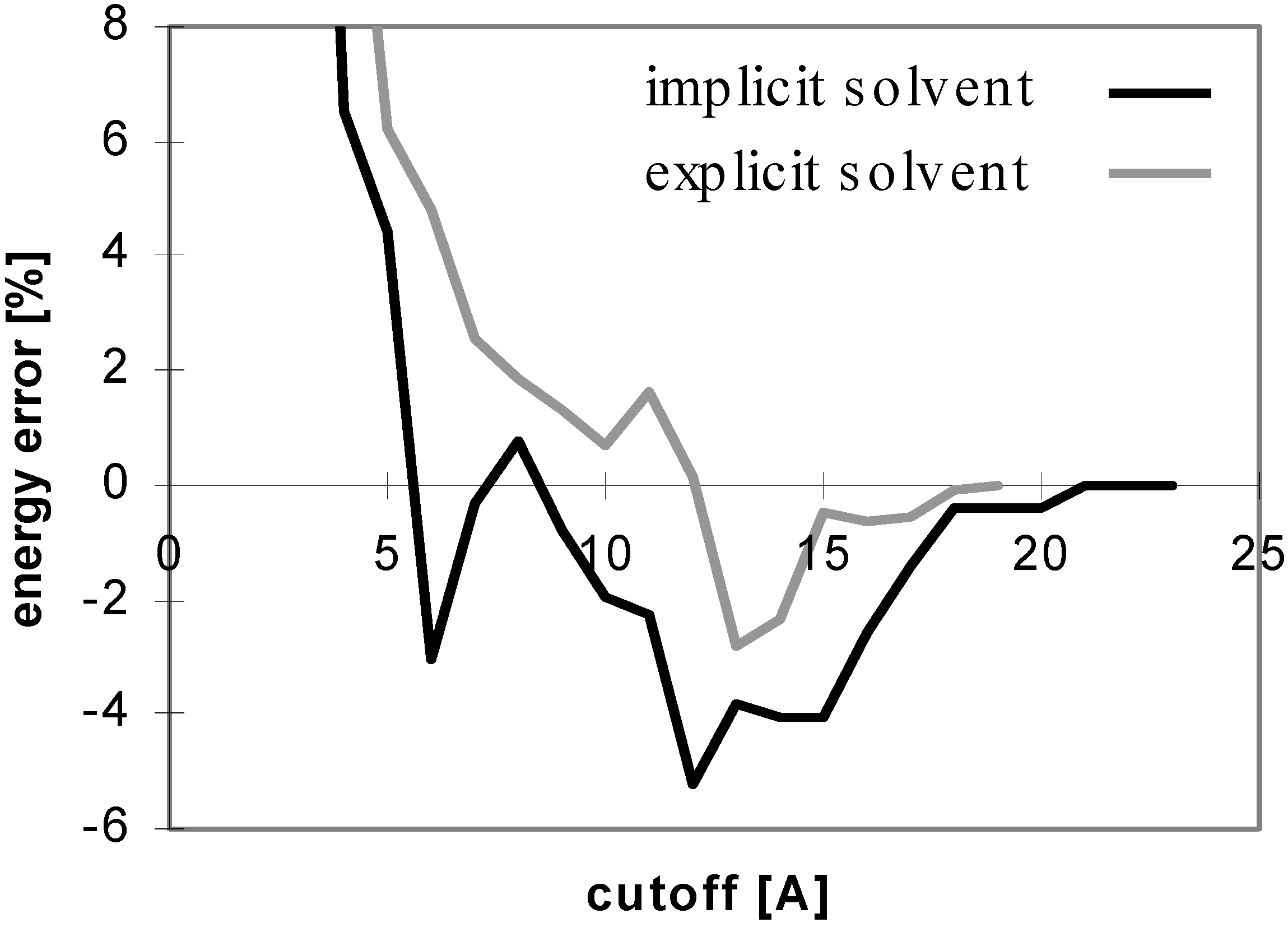
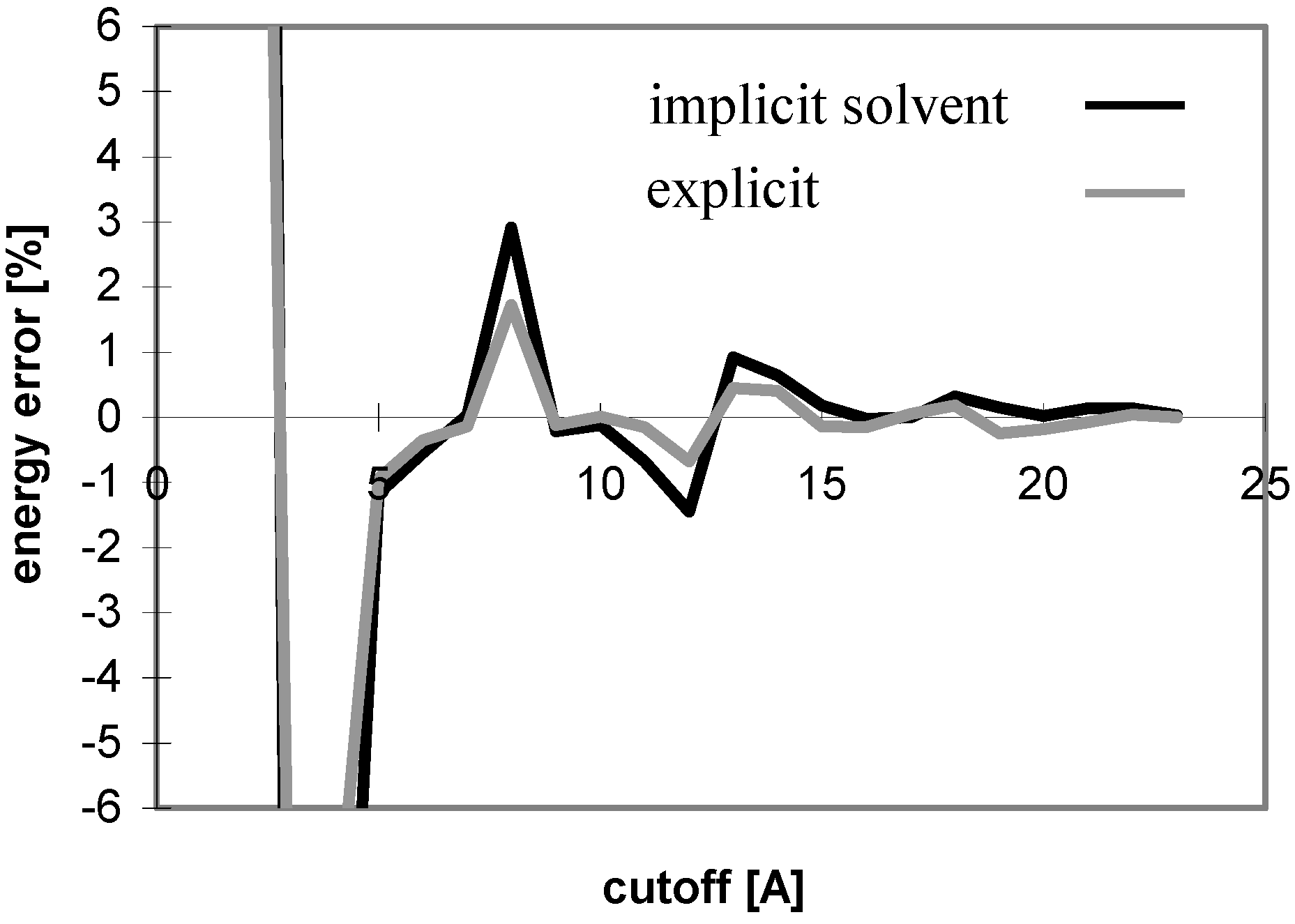
The results obtained for peptides and proteins in explicit solvent are not dramatically different from what we discussed above. A notable difference is seen for the convergence of E
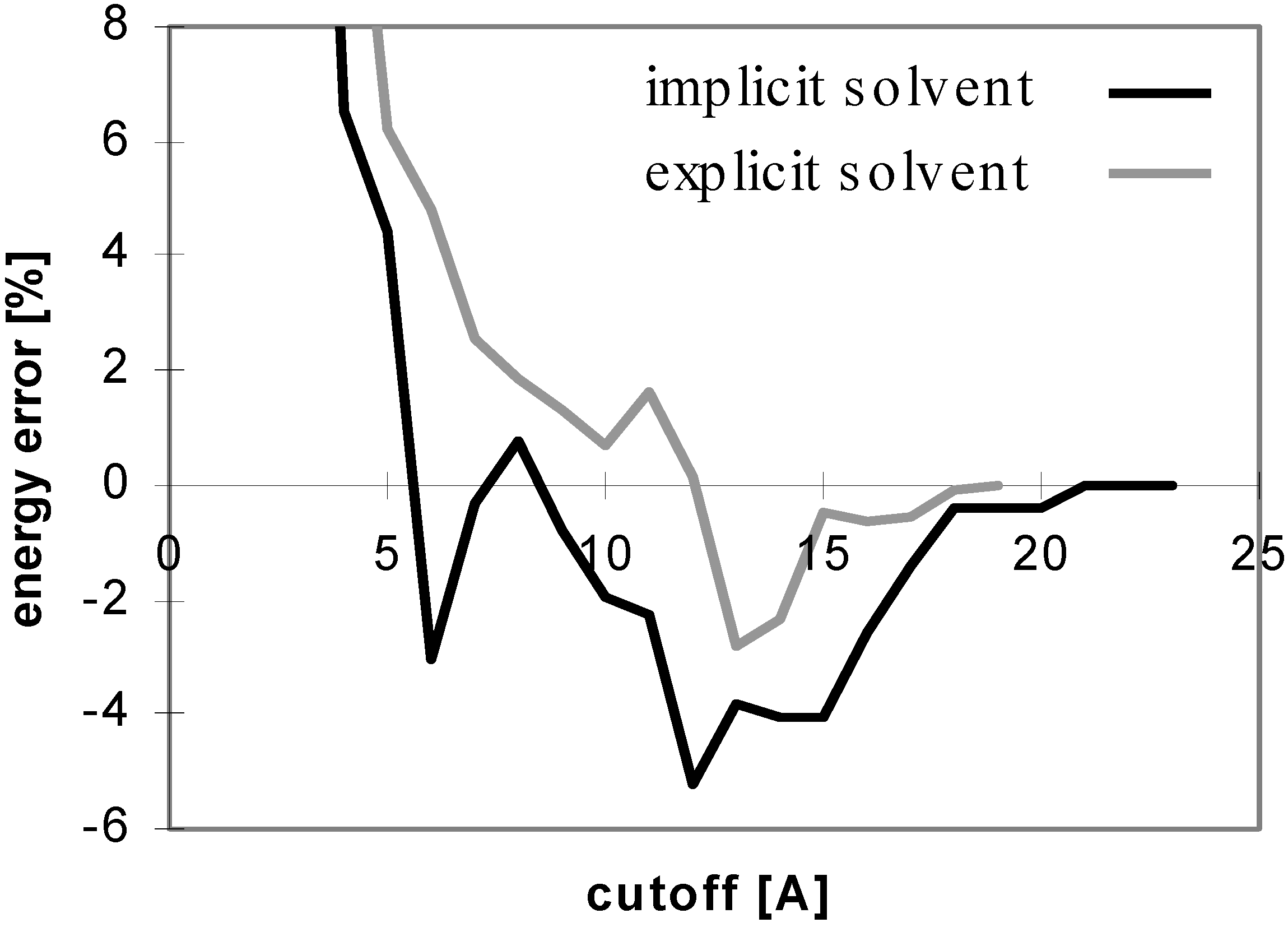
el with the cutoff method where the curve does not oscillate (see
Fig 4), whereas it does in implicit solvent. This is attributed to the coupling of the changes of the solute-solute and solute-solvent contributions to the E
el value with the change of the corresponding solvent-solvent contribution. When only the solute-solute and solute-solvent contributions are included in the E
el value, the curve becomes similar to that in implicit solvent (see
Fig 5).
Figure 4.
Dependency of energy error on cutoff for peptide P2 in explicit solvent. For description of energy error see
Figure 1.
Figure 4.
Dependency of energy error on cutoff for peptide P2 in explicit solvent. For description of energy error see
Figure 1.
Figure 5.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent (default value for dielectric multiplicative constant of 1.0) and explicit solvent (without including solvent-solvent interactions) for peptide P2.
Figure 5.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent (default value for dielectric multiplicative constant of 1.0) and explicit solvent (without including solvent-solvent interactions) for peptide P2.
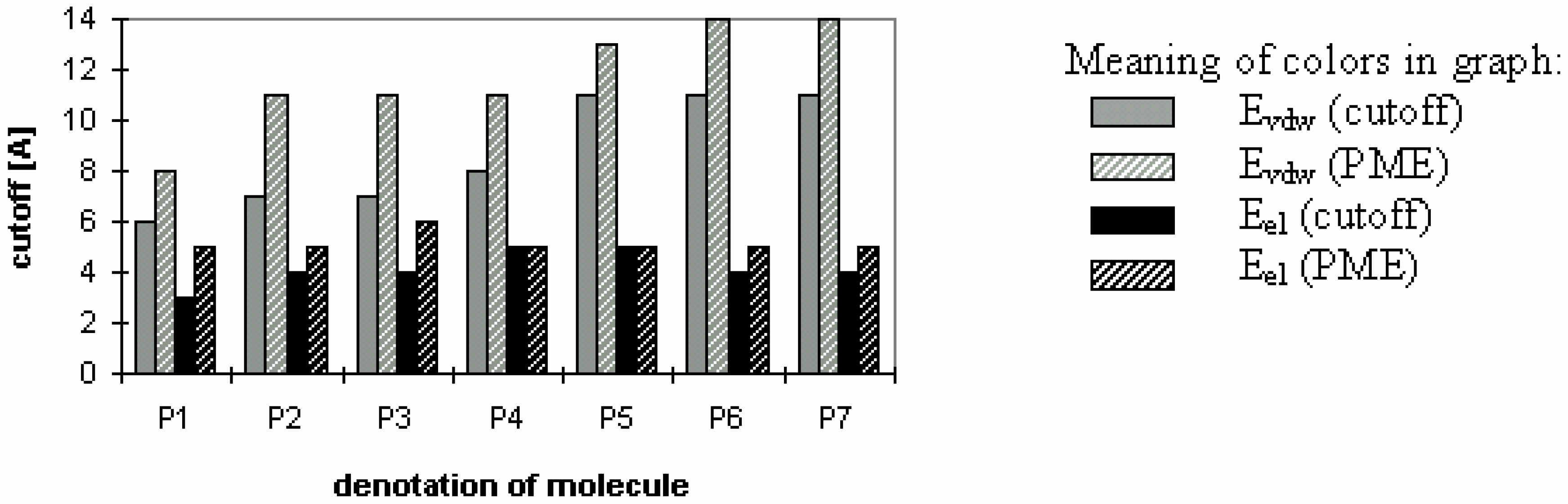
Table 6.
Final energies (kcal/mol) calculated using cutoff E
f(cutoff) and PME E
f(PME) methods. The relative difference of absolute energies δE
f (%) was calculated using equation:
Table 6.
Final energies (kcal/mol) calculated using cutoff Ef(cutoff) and PME Ef(PME) methods. The relative difference of absolute energies δEf (%) was calculated using equation:
| Denotationof molecule | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P2 expl.solv. |
|---|
| Efvdw(cutoff) | -13,99 | -132,37 | -198,34 | -337,22 | -516,44 | -3110,03 | -2851,67 | 3994,22 |
| Efvdw(PME) | -13,98 | -132,31 | -198,13 | -336,92 | -516,26 | -3109,52 | -2852,28 | 3878,69 |
| δ Efvdw | 0,04 | 0,02 | 0,05 | 0,04 | 0,02 | 0,01 | 0,01 | 1,47 |
| Efel(cutoff) | -29,34 | -137,23 | -153,96 | -261,26 | -517,09 | -1887,12 | -1682,18 | -8479,32 |
| Efel(PME) | -98,11 | -422,17 | -500,32 | -762,80 | -1642,17 | -5535,22 | -5130,14 | -8162,36 |
| δ Efel | 53,96 | 50,94 | 52,94 | 48,98 | 52,10 | 49,15 | 50,61 | 1,90 |
Oligonucleotides
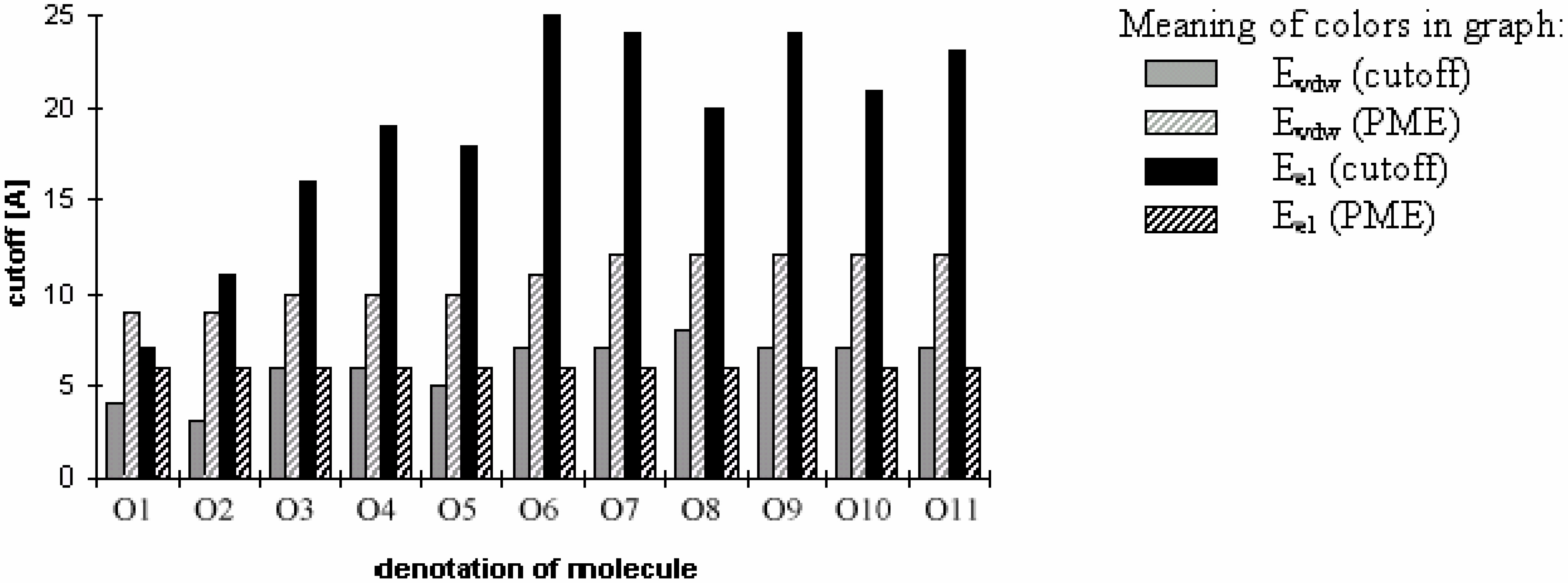
Eleven molecules belonging to this class were studied (see
Table 7). Note that the absolute values of E
fvdw in the explicit solvent are similar for the cutoff and PME methods but the values of E
fel calculated by using PME are markedly larger than the corresponding values calculated by using the cutoff method (see
Table 6). This difference is caused by the absence of explicit water in the grid for PME calculation. The grid includes charges, which simulate long-range interactions in the molecule. If explicit water molecules are present, they shield grid charges and PME provides absolute energies comparable with energies from the cutoff method (see
Table 6). The larger the water box is, the closer to each other the values of E
fel for the two methods are.
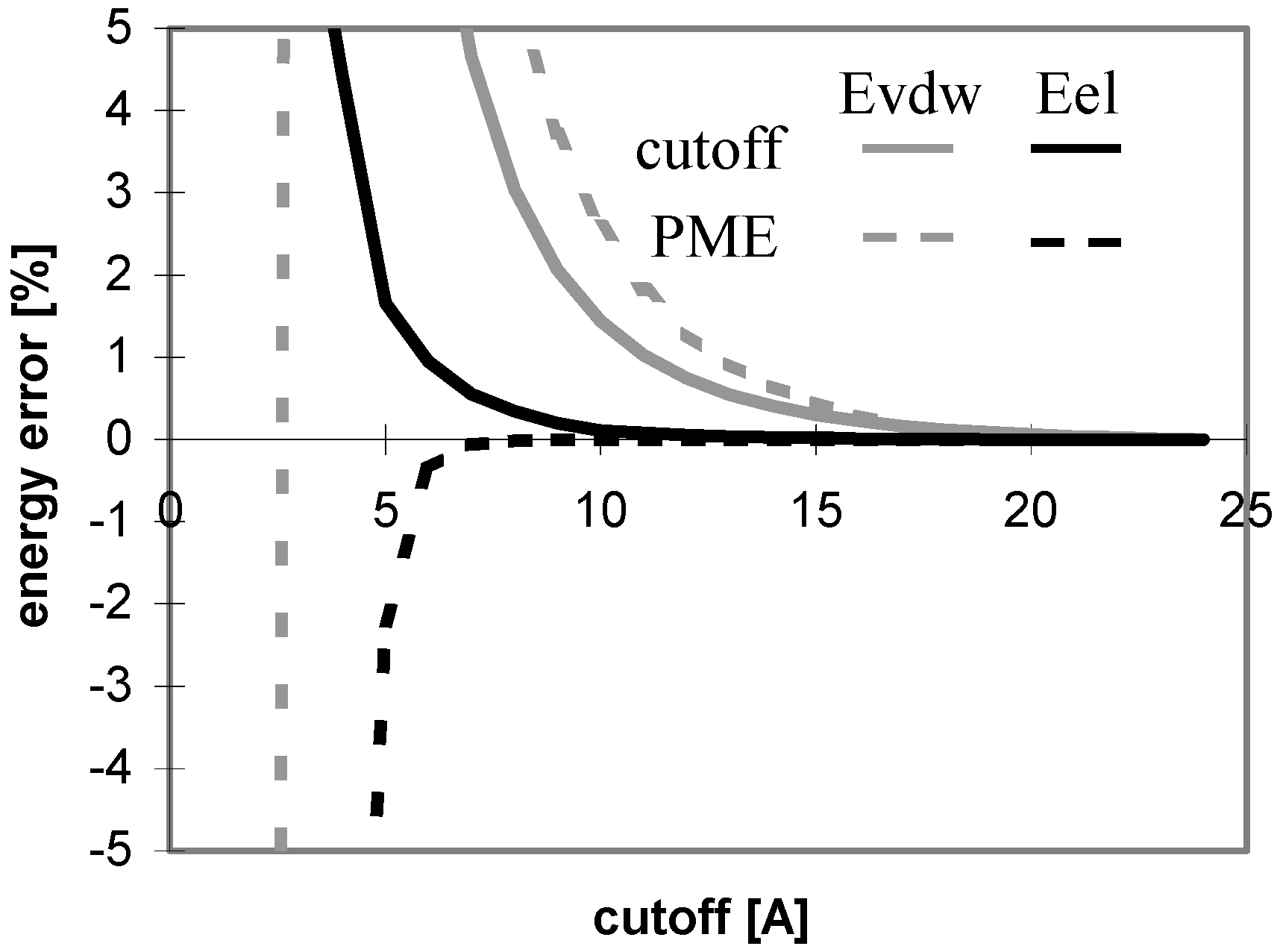
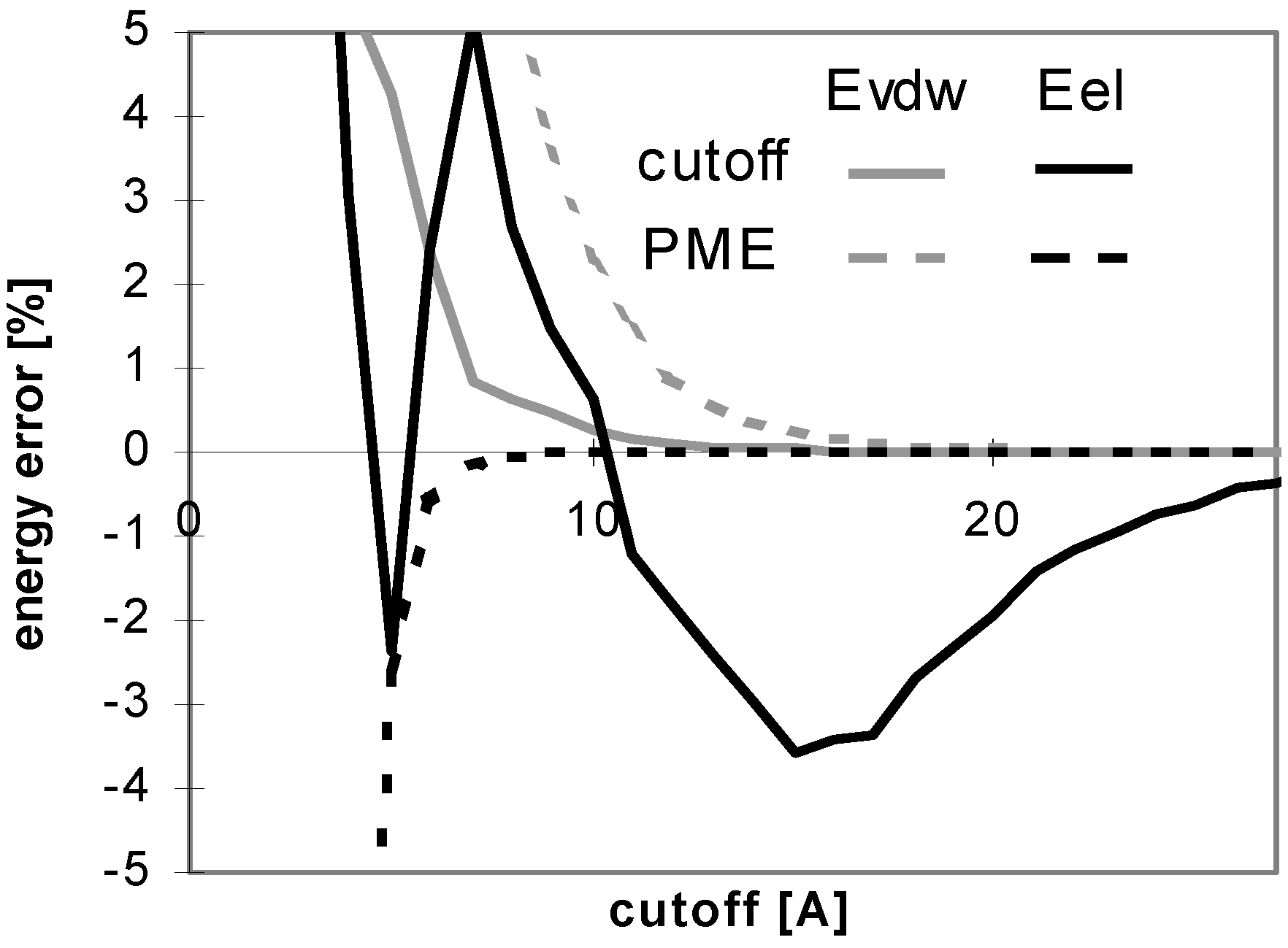
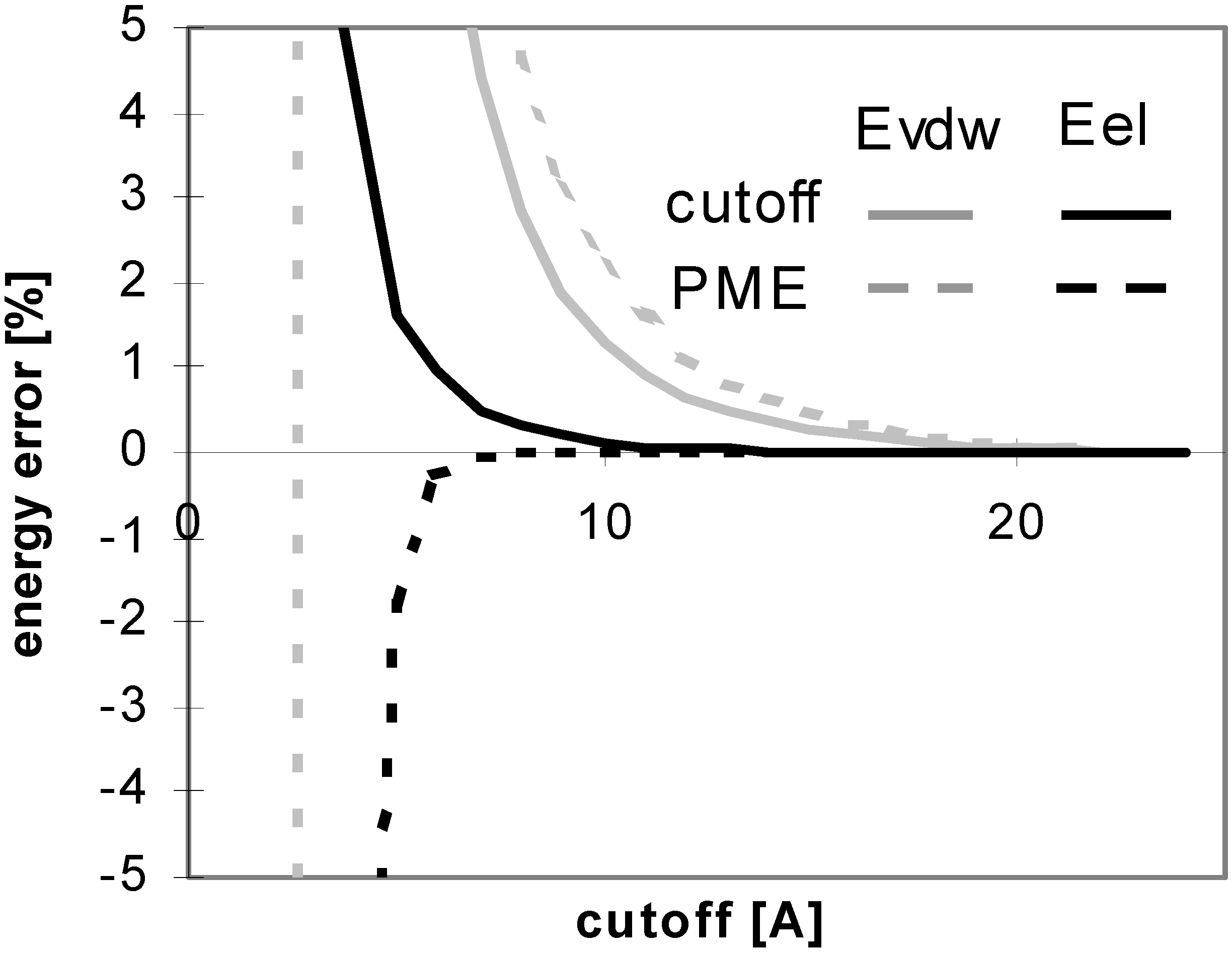
The results of energy calculations for the oligonucleotide O11 are depicted in Fig 6, which shows the convergence of energy to its final value. The general tendencies seen in the results calculated for all of the remaining oligonucleotides are nearly the same (data not shown). The speeds of the convergence of E
vdw for both the cutoff and PME methods and that of E
el for the cutoff method are somewhat dependent on the size, the number of ions, and the 3D structure of the system (see
Fig 7). The convergence of E
el for PME method is independent of the size of the molecule.
Table 7.
Studied oligonucleotides.
Table 7.
Studied oligonucleotides.
| Denotation of molecule | Number of atoms in the system | Base sequence | Number of ions |
|---|
| O1 | 98 | r(AUG) | 2 Na+ |
| O2 | 100 | r(A3) | 2 Na+ |
| O3 | 228 | d(GCGAAGC) | 6 Na+ |
| O4 | 228 | d(GCGAAGC) | 6 Na+ |
| O5 | 228 | d(GCGAAGC) | 6 Na+ |
| O6 | 516 | d(C4G4) | 14 Na+ |
| O7 | 516 | d(C4G4) | 14 Na+ |
| O8 | 728 | r(CCGACGAUAGU)2 | 10 Na+, 5 Mg2+ |
| O9 | 784 | d(GCA4T4GC)2 | 22 Na+ |
| O10 | 784 | d(CGT4A4CG)2 | 22 Na+ |
| O11 | 868 | r(CGCGGCACCGUCC) 2 | 25 Na+ |
Figure 6.
Dependency of energy on cutoff (for oligonucleotide O11). For energy error definition see
Fig. 1.
Figure 6.
Dependency of energy on cutoff (for oligonucleotide O11). For energy error definition see
Fig. 1.
Figure 7.
Smallest values of cutoff for which energy error is smaller then 1% (for oligonucleotides).
Figure 7.
Smallest values of cutoff for which energy error is smaller then 1% (for oligonucleotides).
The convergence of Eel is dramatically slower than that of Evdw for the cutoff method. It means that using different cutoff values for calculating Eel and Evdw should be very efficient in this case; using the two different cutoff values should significantly speed up the computation since Evdw is much more expensive to calculate than the corresponding Eel. The convergence of Evdw for PME method is slightly slower than that of Eel, but the use of different cutoff values for Eel and Evdw would not dramatically affect the speed of computation in this case.
Figure 7 clearly shows that PME is much better than the cutoff method for oligonucleotides. From the figure, one can also clearly see why it was difficult to obtain stable molecular dynamics trajectories on this class of molecules using the cutoff approach to handle electrostatics. One would need to use a very large cutoff value in order to achieve the convergence of the energy. See
Table 8 for a brief summary of these results.
Table 8.
Summary of results for oligonucleotides.
Table 8.
Summary of results for oligonucleotides.
| | Speed of Evdw convergence | Speed of Eel convergence | Evdw / Eel comparison |
|---|
| Cutoff method | fast | very slow | Evdw significantly faster |
| PME method | average speed | very fast | Eel faster |
| Cutoff / PME methods | Cutoff method is faster | PME method is markedly faster | PME method is substantially better |
Figure 8.
Dependency of energy error on cutoff for oligonucleotide O4 in explicit solvent. For description of energy error see
Figure 1.
Figure 8.
Dependency of energy error on cutoff for oligonucleotide O4 in explicit solvent. For description of energy error see
Figure 1.
The results obtained for oligonucleotides in explicit solvent are similar to those discussed above. As seen for proteins and peptides, for oligonucleotides the notable difference also exists in the curve of the convergence of E
el for the cutoff method (see
Fig 8). The reason for this difference is the same as that for proteins,
i.e. due to the coupling with the change of the additional solvent-solvent contribution to the E
el value. When the solvent-solvent contribution is excluded from the E
el calculation, the curve becomes similar to that in implicit solvent (see
Fig 9).
Figure 9.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent and explicit solvent (without including solvent-solvent interactions) for oligonucleotide O4.
Figure 9.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent and explicit solvent (without including solvent-solvent interactions) for oligonucleotide O4.
The same as observed for the absolute energies of the peptides and proteins (see last paragraph of the "Proteins and peptides" section) apply also for the absolute energies of the studied oligonucleotides.
Nucleotide-sugars
Three conformers (denoted by N1, N2, and N3) of GTP-fucose (80 atoms, 10 Cl-, 9 Na+, 3 K+) belonging to this class were calculated.
We have selected systems with different space locations of the ions, so the overall shape of the molecule itself as well as of the entire system was different. The basic geometrical parameters are shown in
Table 9.
Table 9.
Selected geometrical parameters of three different conformations of the nucleotide-sugar.
Table 9.
Selected geometrical parameters of three different conformations of the nucleotide-sugar.
| | N1 | N2 | N3 |
|---|
| Size of molecule* [Å] | 7,68 | 7,5 | 9,56 |
| Overall size of the entire system** [Å] | 3529,88 | 3268,46 | 1056,74 |
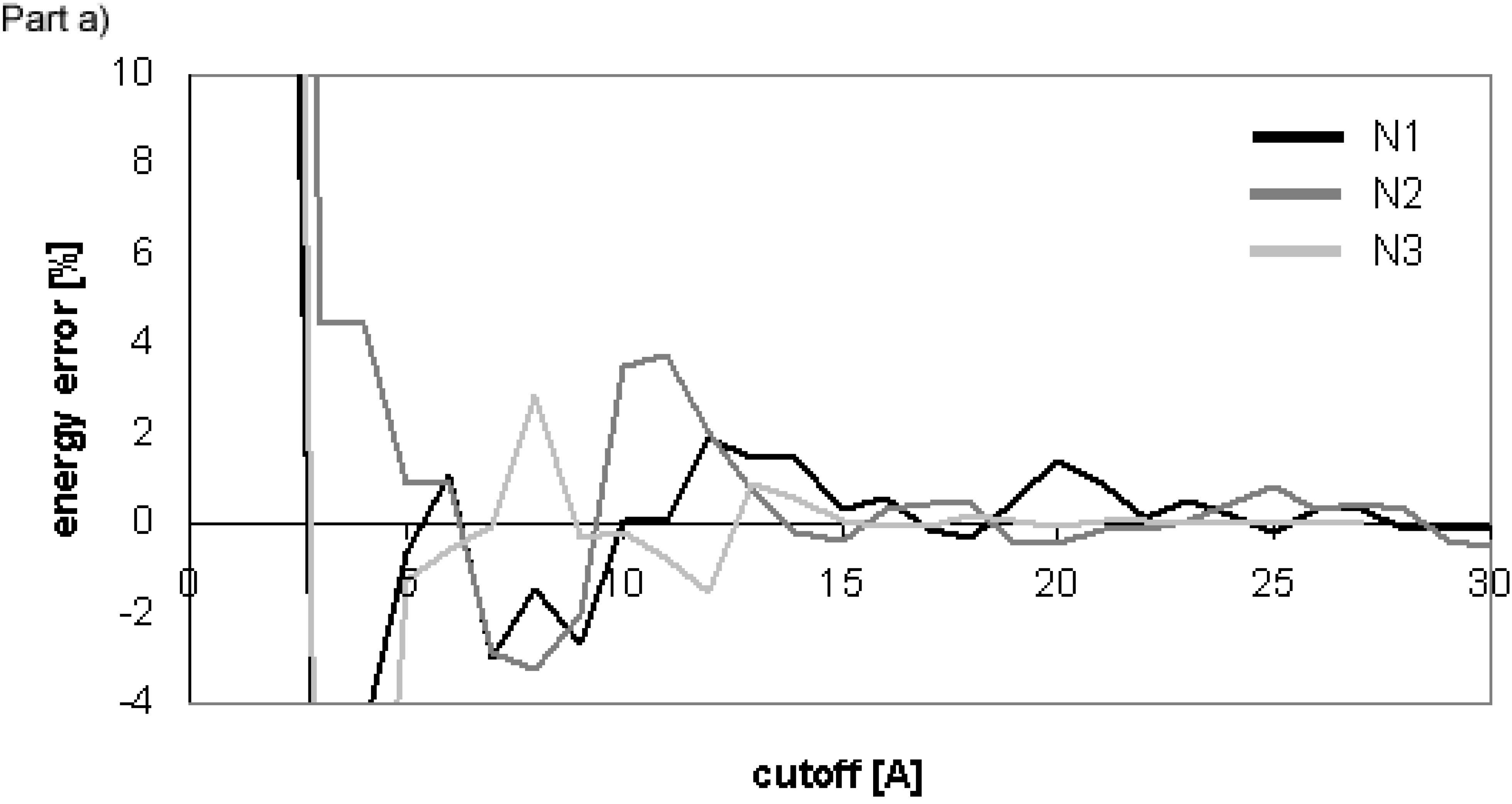
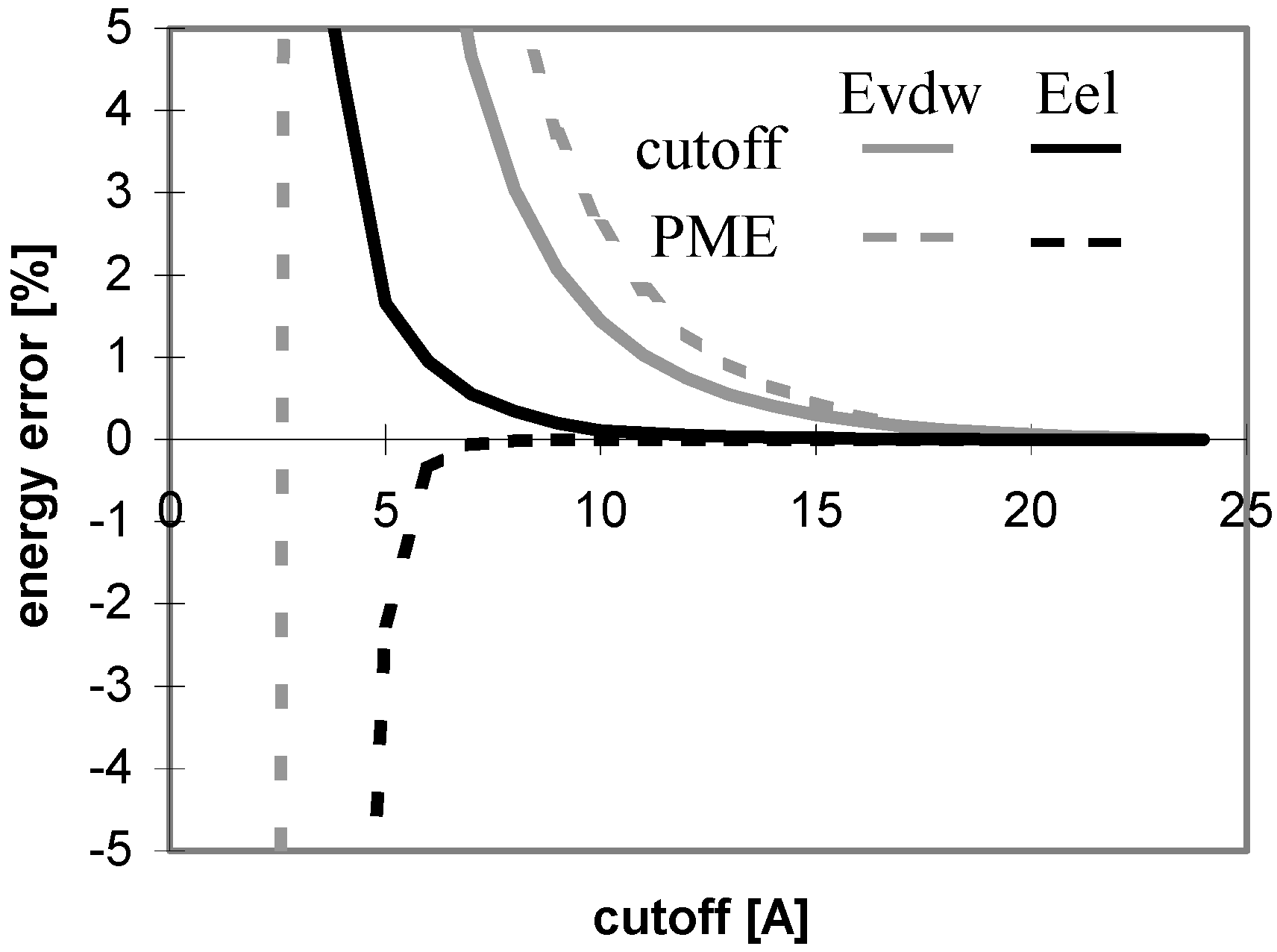
The energy convergence for all these systems is shown in
Fig 10 and
Fig 11. As shown in the figures, the convergence is clearly dependent on the overall shape of the system. The only exception is for van der Waals interactions the speed of the convergence depends also on the size of molecule.
Figure 10.
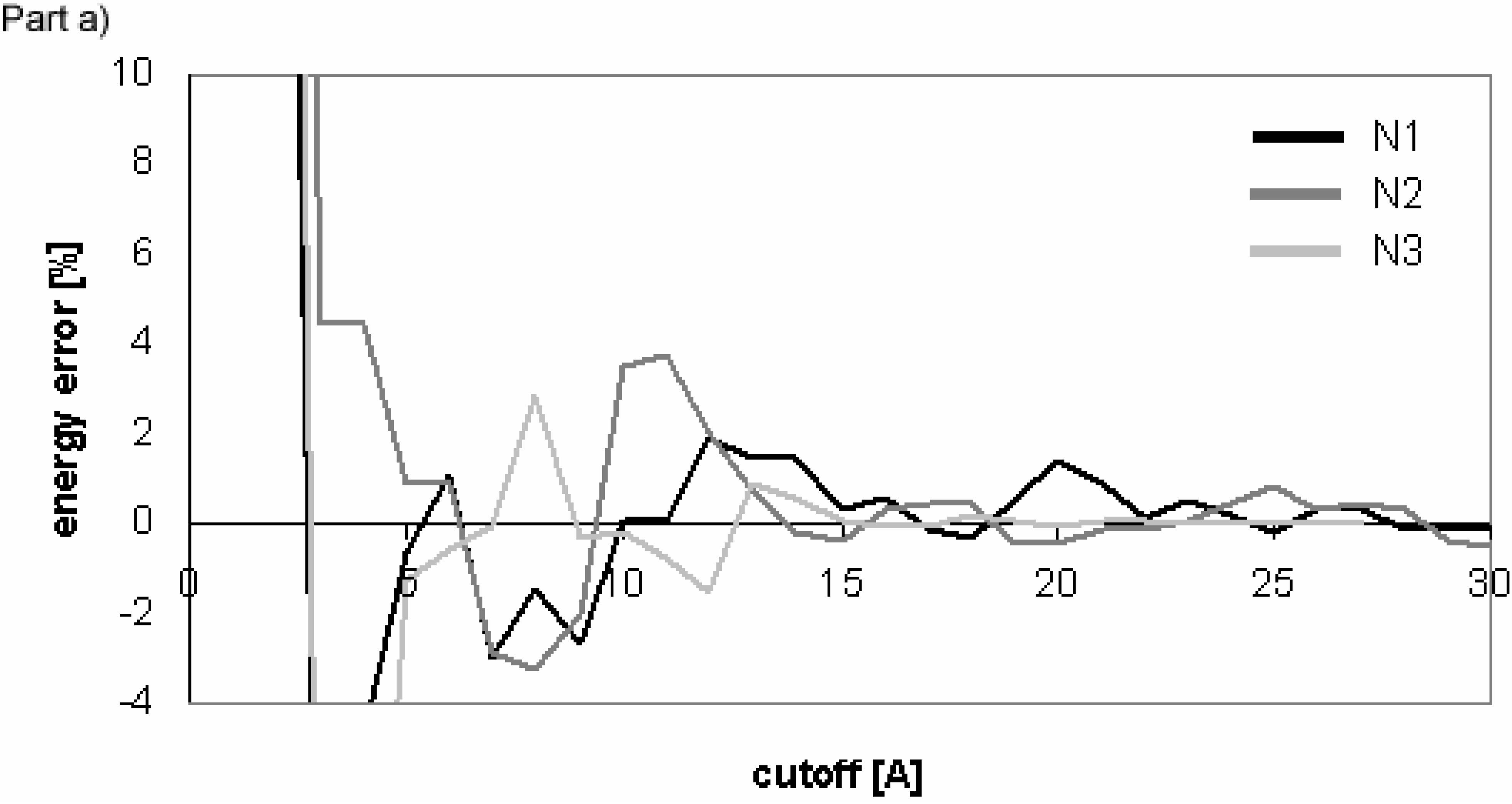
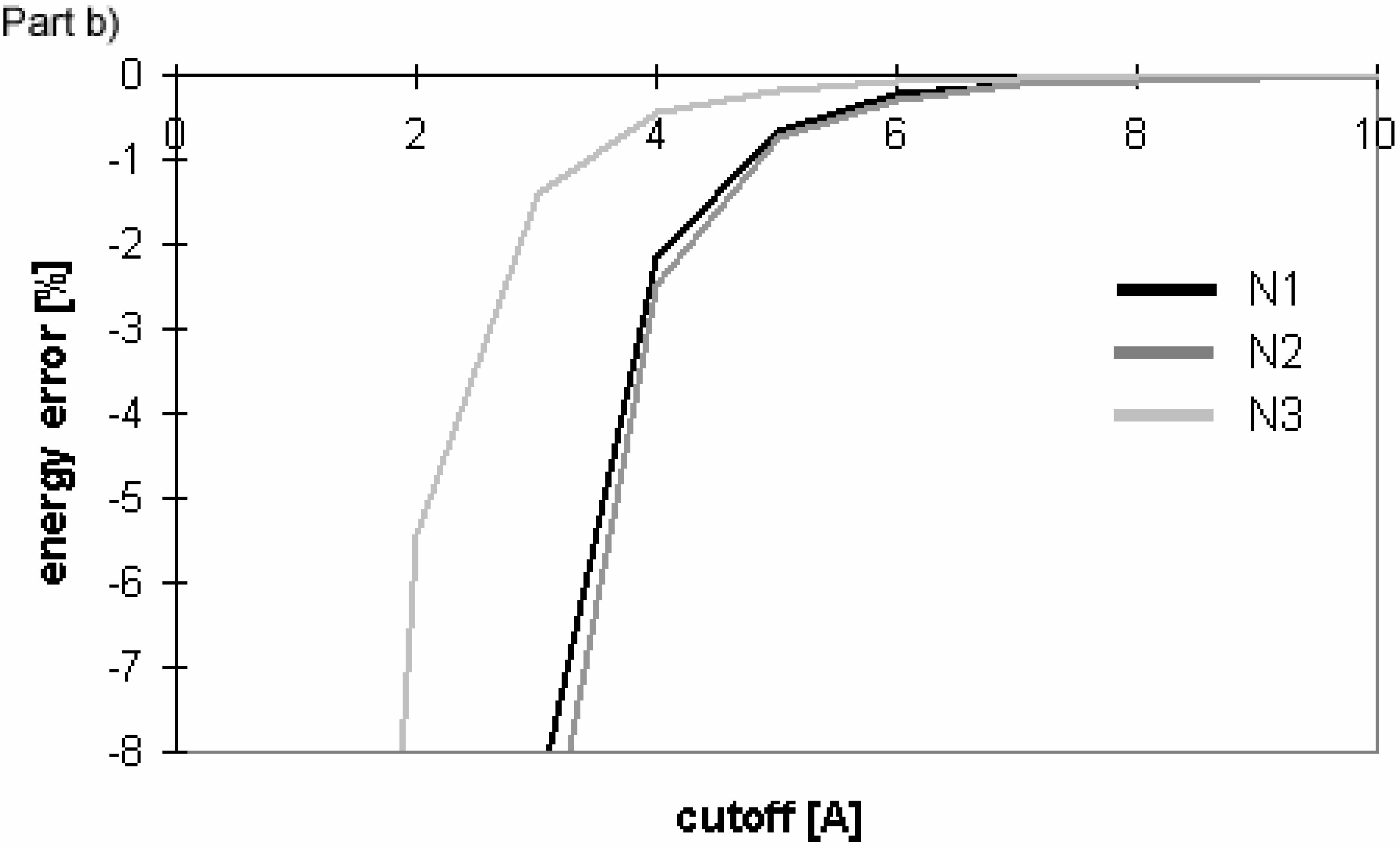
Dependency of energy error on cutoff for conformers of nucleotide-sugar. Energy error is calculated for Eel using cutoff method (Part a)) and PME method (Part b)).
Figure 10.
Dependency of energy error on cutoff for conformers of nucleotide-sugar. Energy error is calculated for Eel using cutoff method (Part a)) and PME method (Part b)).
Figure 11.
Dependency of energy error on cutoff for conformers of nucleotide-sugar. Energy error is calculated for Evdw using cutoff method (Part a)) and PME method (Part b)).
Figure 11.
Dependency of energy error on cutoff for conformers of nucleotide-sugar. Energy error is calculated for Evdw using cutoff method (Part a)) and PME method (Part b)).
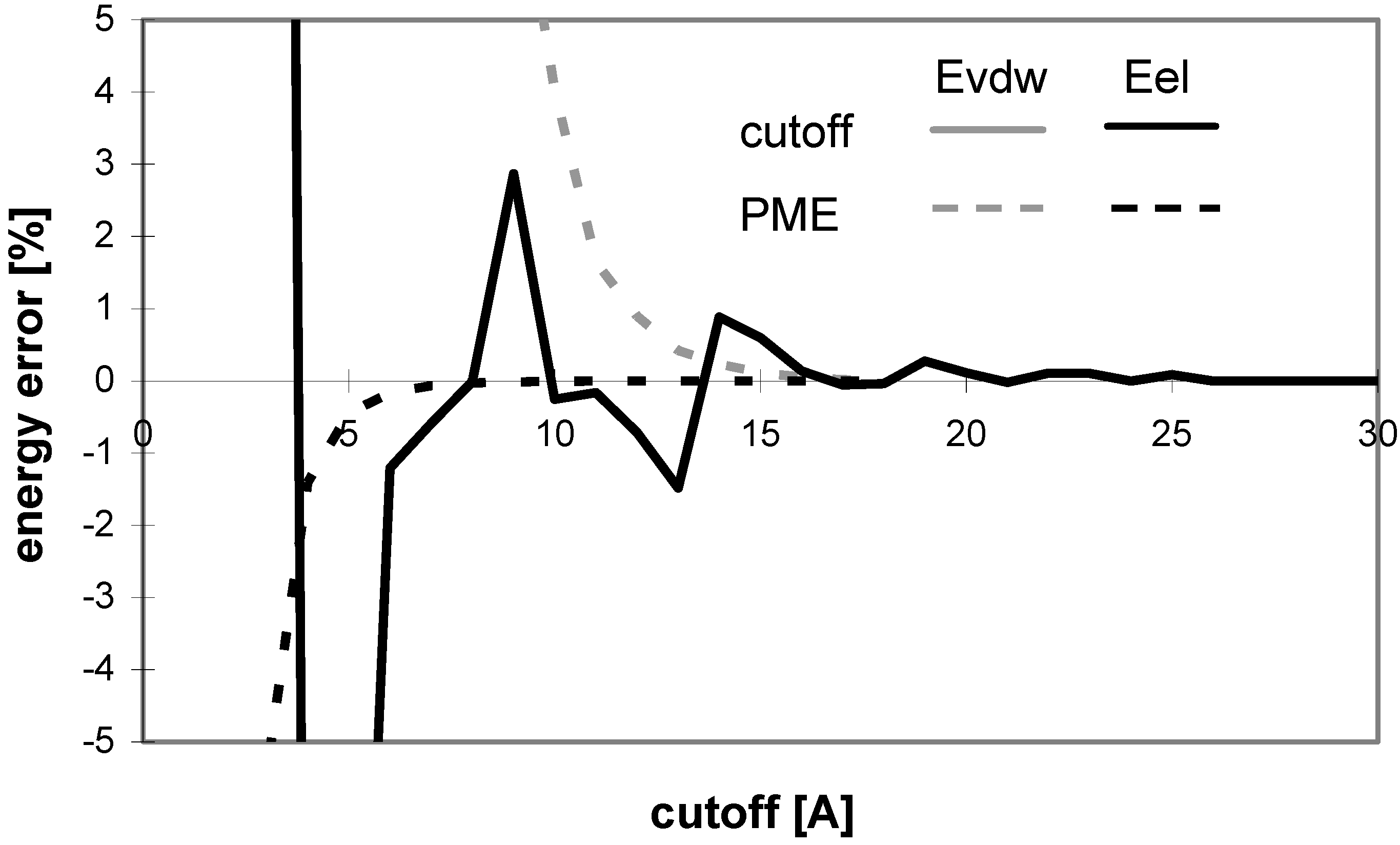
The results of energy calculations for the N3 conformer are pictured in
Fig 12 showing the convergence of the calculated energies to their final values. The results calculated for all of the remaining conformers are similar.
The overall results indicate that for these molecules Evdw converges slowly and Eel very slowly for the cutoff method. The difference between the convergence of Eel and that of Evdw for the nucleotide-sugar is somewhat between the differences for peptides/proteins and for oligonucleotides. The convergence of Eel within the cutoff method is slightly slower than that of Evdw. It means that using different cutoff values for Eel and Evdw would be efficient for the computation, but the speed-up of the computation would not be dramatic as the difference between the two cutoff values is small.
Within PME method, E
vdw converges slowly and E
el very quickly, which is similar to the case of oligonucleotides. The convergence of E
vdw is markedly slower than that of E
el, indicating that the use of two different cutoff values for E
el and E
vdw would be efficient for the computation, but the speed-up
Figure 12.
Dependency of energy on cutoff for (nucleotide-sugar N3). For energy error definition see
Fig. 1.
Figure 12.
Dependency of energy on cutoff for (nucleotide-sugar N3). For energy error definition see
Fig. 1.
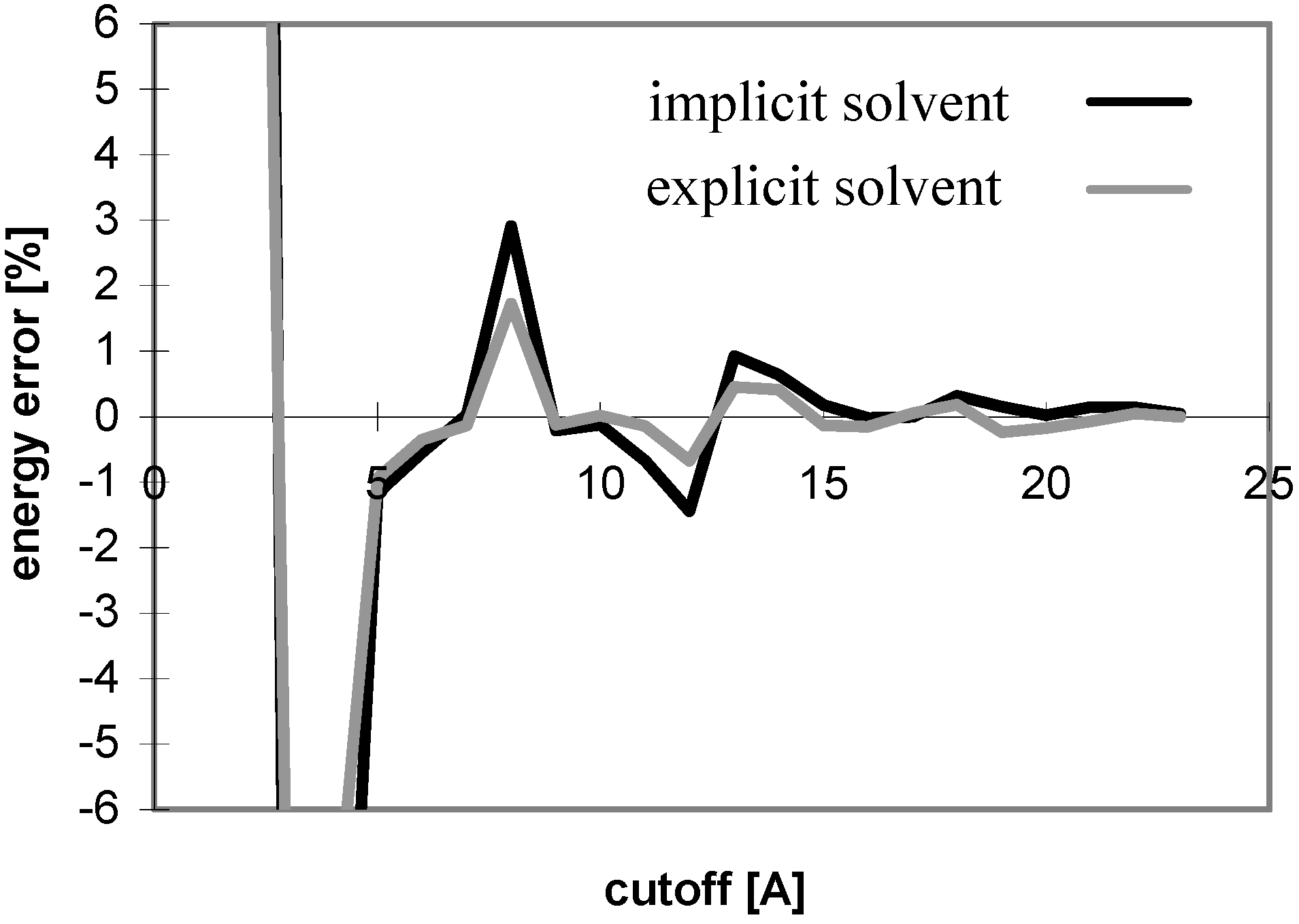
Figure 13.
Dependency of energy error on cutoff for nucleotide-sugar N3 in explicit solvent
Figure 13.
Dependency of energy error on cutoff for nucleotide-sugar N3 in explicit solvent
of the computation would be insignificant, because the computation of E
el is relatively faster than that of E
vdw.
Fig 12 clearly suggests that for nucleotide-sugars, in terms of the energy convergence, the performance of PME method is generally better than the cutoff method. However, when a very high-accuracy is desired for the energy evaluation, the cutoff method can also be used and may even be more efficient. Whereas the cutoff method requires a large cutoff value for E
el, PME needs a similarly large cutoff value for E
vdw whose calculation is more expensive than the E
el calculation. See
Table 10 for a brief summary of these results.
Table 10.
Summary of results for nucleotide-sugars.
Table 10.
Summary of results for nucleotide-sugars.
| | Speed of Evdw convergence | Speed of Eel convergence | Evdw / Eel comparison |
|---|
| Cutoff method | average speed | slow | Evdw faster |
| PME method | slow | fast | Eel significantly faster |
| Cutoff / PME methods | Cutoff method is faster | PME method is markedly faster | PME method is better |
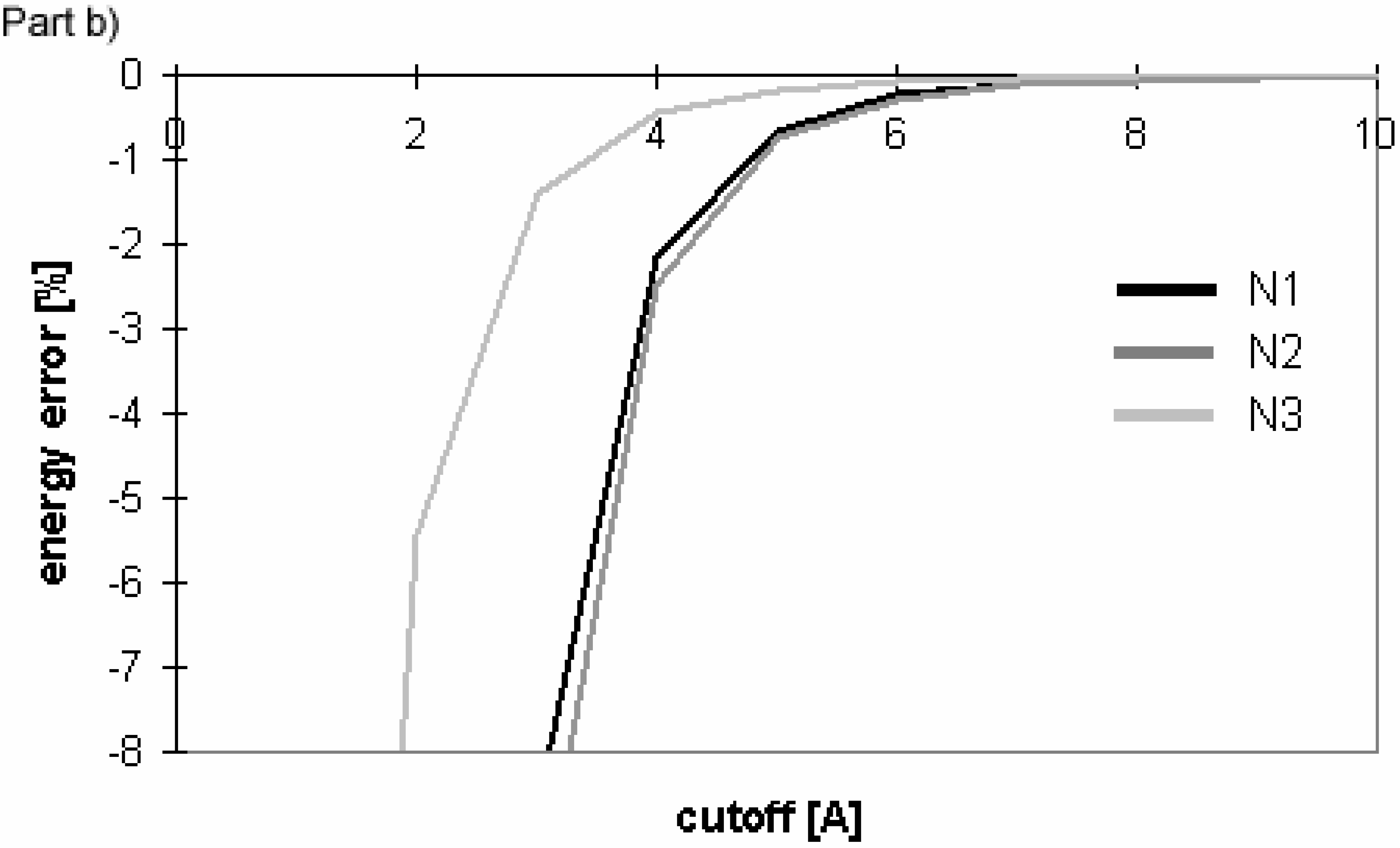
The results obtained for the nucleotide-sugar in explicit solvent well correspond with those obtained for peptides and oligonucleotides (see
Fig 13 and
Fig 14). The same applies for the cutoff and PME absolute energies (data not shown).
Figure 14.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent and explicit solvent (without including solvent-solvent interactions) for nucleotide sugar N3.
Figure 14.
Dependency of electrostatic energy (calculated using cutoff method) on cutoff for implicit solvent and explicit solvent (without including solvent-solvent interactions) for nucleotide sugar N3.
Possibilities to improve the convergence performance by changing parameters
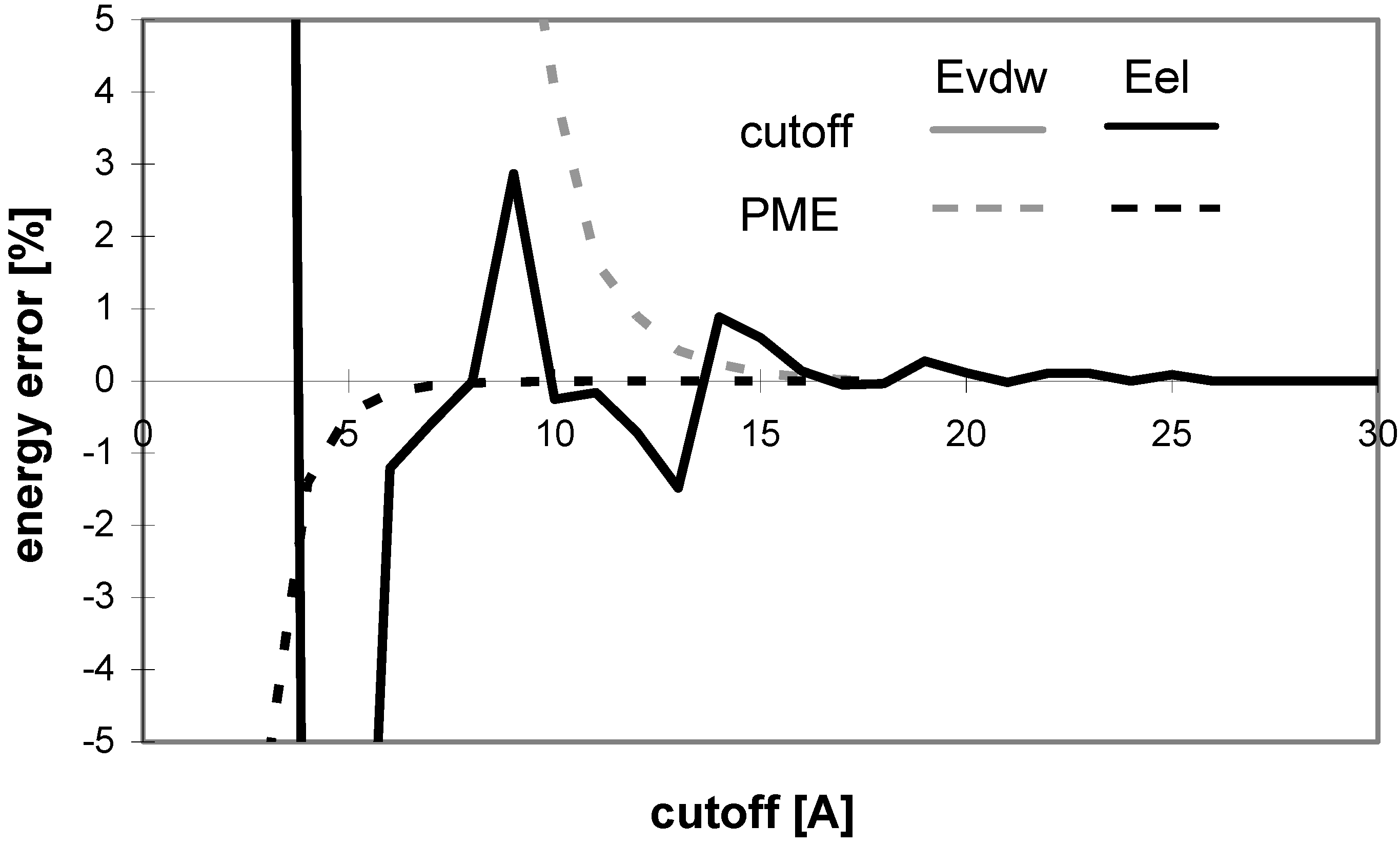
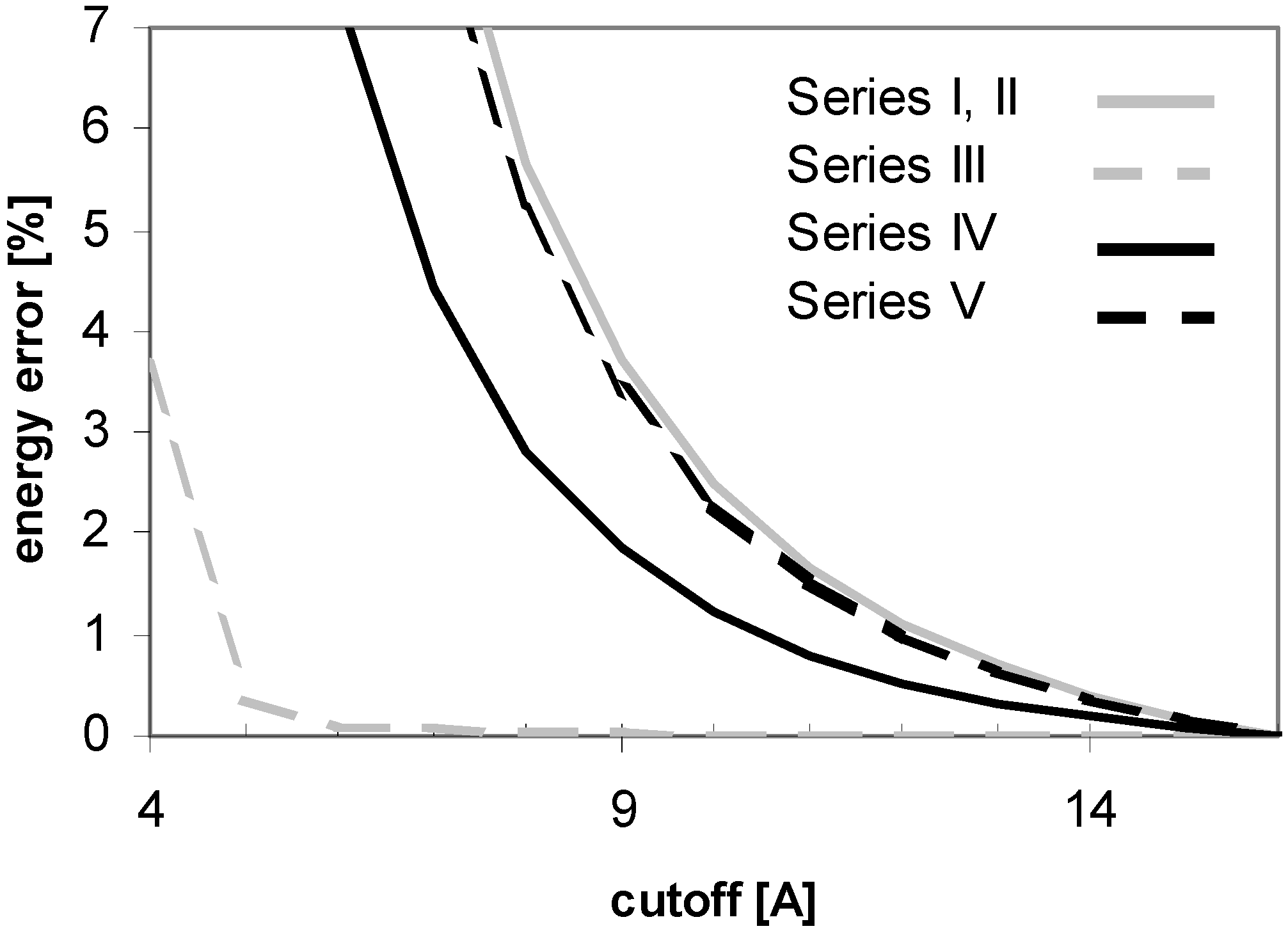
The above results were obtained using the version 5.0 of AMBER. There are two problematic issues that can be improved either by changing PME parameter values for the calculations or by using a more recent version of AMBER (6.0 or 7.0) which uses more parameters to control the computation and also different default PME parameter values. The first issue is associated with the observation that the PME energies calculated with very small cutoff values are dramatically different from the corresponding final energies converged at the large cutoff values. However, this problem can easily be solved by increasing the spline order and/or reducing the grid spacing. We observed a much improved convergence performance by just reducing the grid spacing (data not shown). On the other hand, actually one does not need to worry about the dramatically different energies calculated by using very small cutoff values because no one would really use such a small cutoff value in practical calculations.
The second problem, which looks more serious, is associated with the observation that the convergence of vdW energy is different for the calculations with the cutoff and PME methods. In principle, the convergence of vdW energy should be in both cases exactly the same because vdW evaluation is not dependent on using PME. As far as we have known [
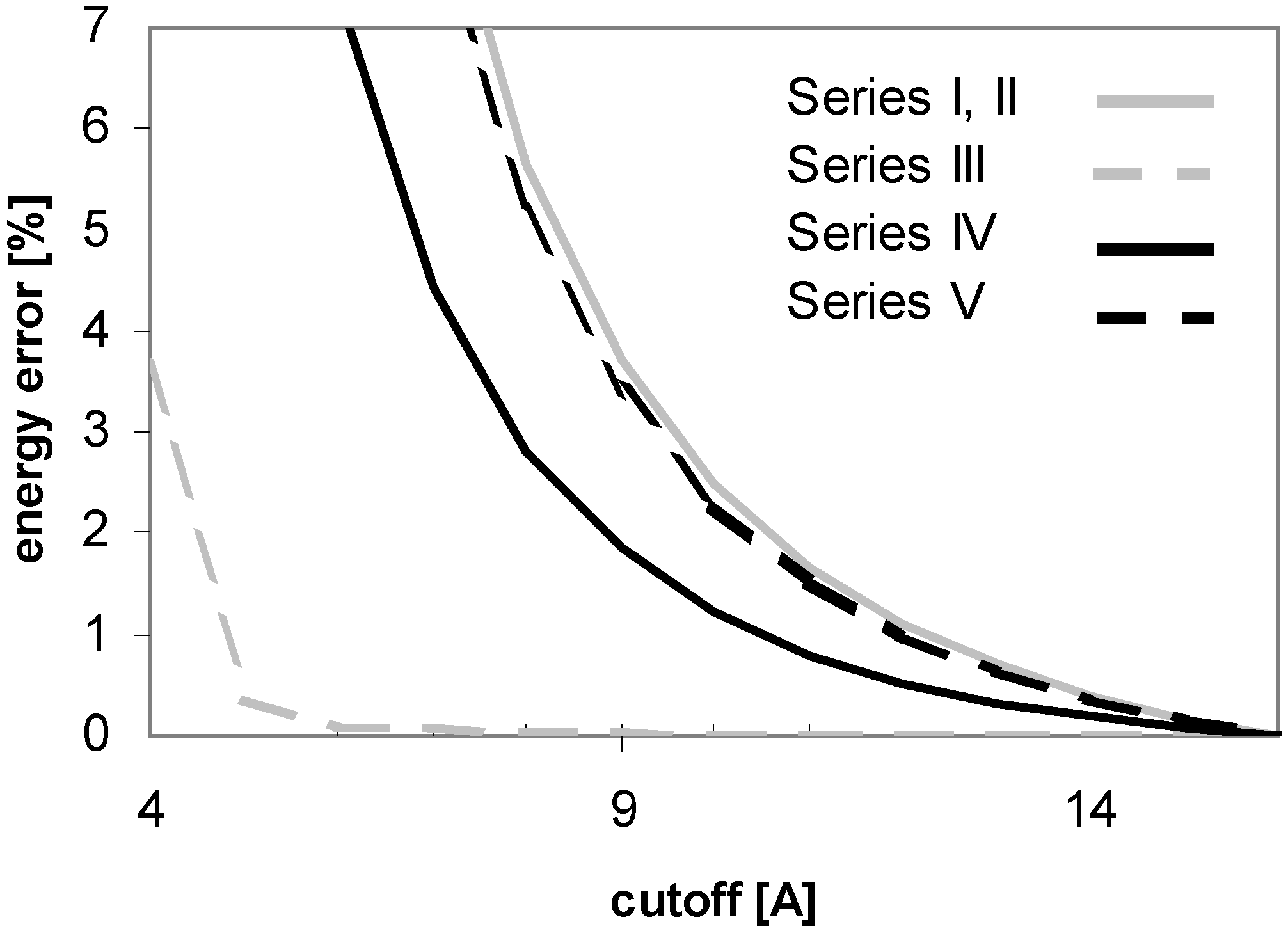
33], the PME-based calculations implemented in AMBER is associated with an "atom based pairlist", in contrary to the "(neutral charge-) group based pairlist" used in the calculations without using PME in AMBER 5.0 or the earlier versions. It is this difference that causes the notable discrepancy between the vdW energy convergencies. However, when using AMBER 6.0 or 7.0, the additional homogeneous long-range correction to the vdW may optionally be enabled (VDWMETH=1), which includes a bulk density vdW correction. This setup improves the vdW convergence considerably, as shown in
Fig. 15.
Figure 15.
Dependency of energy error on cutoff (for peptide P2) as calculated by AMBER 5.0 and 6.0 with different setup. Denotation: Series I: AMBER 6.0 - cutoff method (USE_PME=0, EEDMETH=4); Series II: Amber 6.0 - PME, Evdw without correction (USE_PME=1, EEDMETH=1, VDWMETH=0); Series III: Amber 6.0 - PME, Evdw with correction (USE_PME=1, EEDMETH=1, VDWMETH=1); Series IV: Amber 5.0 - cutoff method; Series V: Amber 5.0 - PME method.
Figure 15.
Dependency of energy error on cutoff (for peptide P2) as calculated by AMBER 5.0 and 6.0 with different setup. Denotation: Series I: AMBER 6.0 - cutoff method (USE_PME=0, EEDMETH=4); Series II: Amber 6.0 - PME, Evdw without correction (USE_PME=1, EEDMETH=1, VDWMETH=0); Series III: Amber 6.0 - PME, Evdw with correction (USE_PME=1, EEDMETH=1, VDWMETH=1); Series IV: Amber 5.0 - cutoff method; Series V: Amber 5.0 - PME method.
A detailed comparison of the results indeed shows the same vdW energy convergences for the PME and cutoff methods implemented in AMBER 6.0 and the PME method implemented in AMBER 5.0. It appears that the calculation with the cutoff method by using AMBER 5.0 without the additional vdW energy correction converges faster than the corresponding calculation by using AMBER 6/7 with the additional vdW energy correction. For PME, on the other hand, AMBER 6/7 versions are better tuned than AMBER 5.0.
Finally, it should be mentioned that we only examined single-point energies in discussion of the convergence of the energy calculations. Obviously, these energetic results depend on conformation. To exclude this drawback, we were interested rather in relative energies, or, better to say, in energy tendencies. Our conclusions are supported by the fact that for all molecules within one general type identical trends were found. We also note here that this kind of inspection is more detailed that, for example, a molecular dynamics (MD) simulation would be. In the case of MD simulation many energy calculations must be performed and the errors may mutually be canceled, so the final observation may somewhat be deformed. Therefore, we believe that our approach to compare cutoff and PME is reasonable.
Conclusions
Long-range electrostatic and van der Waals interactions still belong to the most problematic part of energy calculation within empirical force field approach of computational chemistry, mainly within molecular mechanics and dynamics. Due to time complexity it is usually not possible to include all interactions into calculations and some approximations must be used. Our effort with this paper has been to compare two most frequently used approximations to handle long-range electrostatic and van der Waals interactions, cutoff, and PME methods. Altogether, 19 molecules have been calculated, for one of them we considered three different conformations. These calculated molecules include peptides, proteins, oligonucleotides, and nucleotide-sugar. The main program package used is AMBER, versions 5.0, 6.0, and 7.0. Some results were obtained also by software written by us.
First, we have compared the time complexities of the calculations of the electrostatic and van der Waals energy components. We have shown by theoretical considerations and also by practical computations that the van der Waals energy component is roughly about 3 times more expensive to calculate than the corresponding electrostatic component. Thus, it is computationally more efficient to use different cutoff values for calculating the electrostatic and van der Waals interactions, particularly for the cases when the van der Waals component requires a lower cutoff value. We have also compared the overall speeds of the calculations with the cutoff and PME methods and concluded that the calculation with the PME method is generally slower, and in some cases is substantially slower, which is consistent with previous results in literature.
Then, we have examined the electrostatic and van der Waals energy convergence to their final values for different molecules, for different cutoff values, and for different methods. This has been performed for both the implicitly and explicitly solvated systems; the implicit solvent is accounted for through the use of a distance-dependent dielectric constant. The borderline cutoff value is the maximum cutoff value including all interactions for the cutoff method or one half of the shortest edge of the PME box for the PME method. The qualitative results are basically the same for both the implicitly and explicitly solvated systems. We have also compared the absolute electrostatic energies calculated by using the cutoff and PME methods. While these values are very different in the implicitly solvated systems, they become almost identical in the explicitly solvated systems. The reason is that the charges in PME grid are not shielded by water molecules in the implicit solvent and, therefore, create an artificial energy term that is not used with the explicit solvent. In general, the energy convergence is found to be dependent on the shape of the system. It has also been concluded that van der Waals energy component convergence is strongly dependent on size of the system measured by the number of atoms. The larger the system, the slower the energy convergence.
It has been observed that the energy convergence is dependent on the type of molecule. Specifically, it is different for peptides, oligonucleotides, and the nucleotide-sugar conformations. For example, with both the PME and cutoff methods peptides and proteins exhibit a slower convergence of van der Waals energy than that of Eel. For Evdw, the cutoff method exhibits the faster convergence than PME and, therefore, using the cutoff method, rather than the PME, is preferred for peptides and proteins, at least from the energy point of view. Because Eel is relatively inexpensive to calculate as compared to Evdw, the use of different cutoff values for calculating different energy components is insignificant for peptides and proteins.
The situation is quite different for oligonucleotides, where the convergence of Eel is very slow with the cutoff method and, therefore, we recommend not to use the cutoff method for this class of compounds unless the systems are very small and the used cutoff value can cover the entire system. This is in a good agreement with the literature data leading to a recommendation for using PME exclusively on oligonucleotides. Using the cutoff method on oligonucleotides would require very careful tuning of the cutoff value. On the other hand, if the cutoff method is used, employing two cutoff values (one for Evdw and one for Eel) could significantly save the time of computation.
The situation for the nucleotide-sugar is somewhat in between those for peptides and proteins and for oligonucleotides. The results show that both the PME and cutoff methods may be used for this class of compounds. However, when the cutoff method is used, the cutoff values should carefully be tuned. In this case, using different cutoff values for Evdw and Eel could also save some time of computation. But using the PME appears safer for oligonucleotides.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}