Introduction
Short-range static and/or dynamic disorder is an “undesirable guest”, which affects a considerable number of crystal structures. When some disorder is found in a crystal structure, it is often impossible to obtain sensible geometric features such as bond distance and angles, intermolecular contacts and packing arrangements. Theoretical calculations can then be used to achieve a better interpretation of the electron density from a crystal structure presenting various degrees of structural short-range disorder.
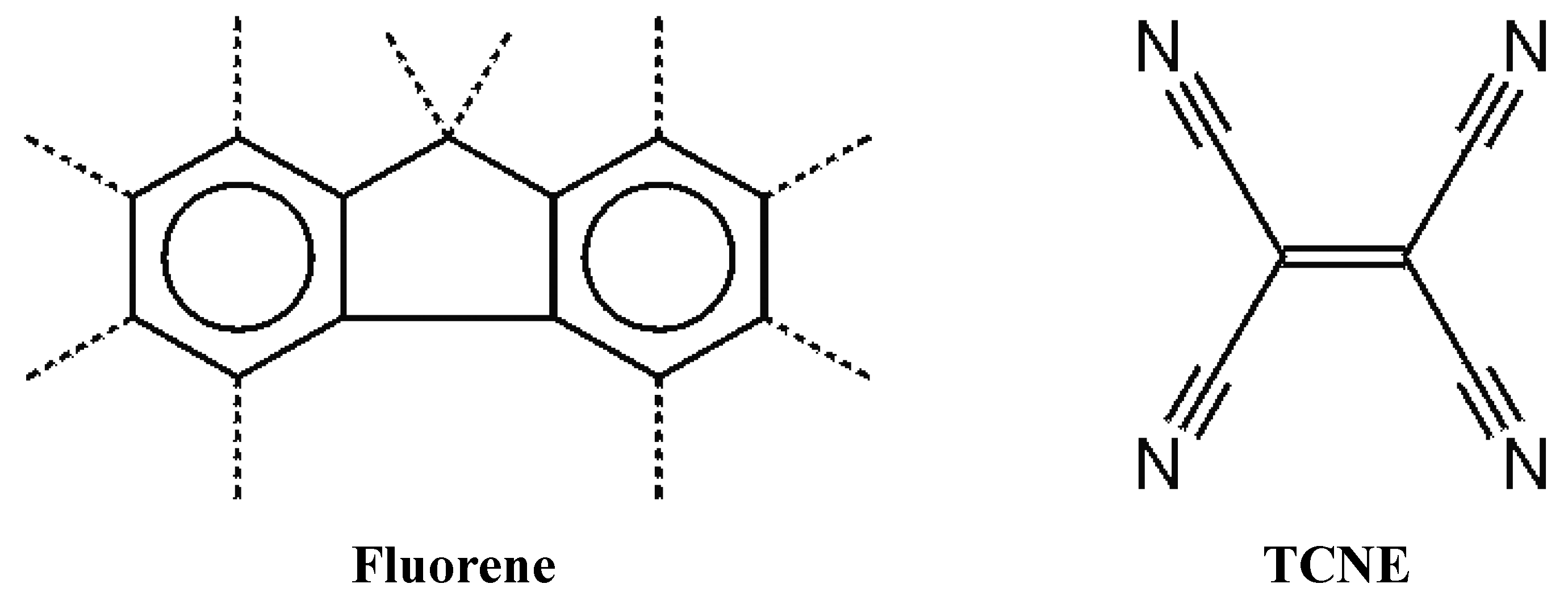
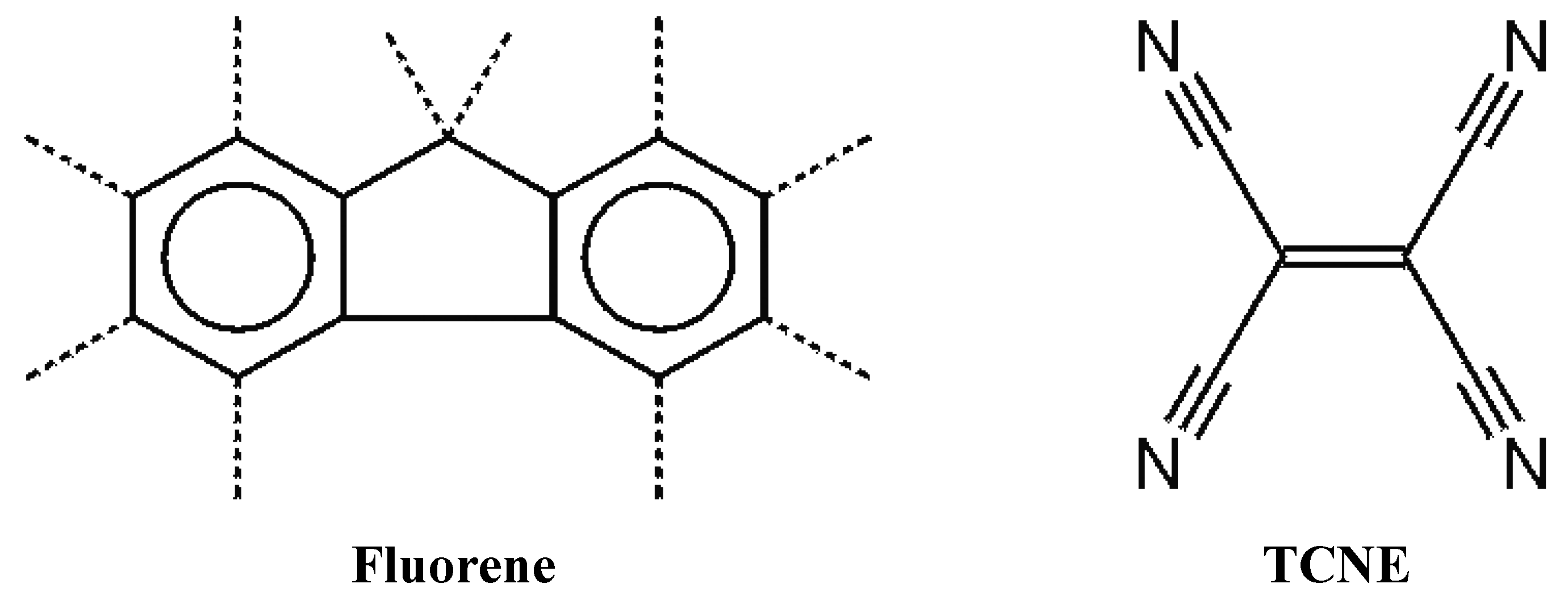
Recently, we decided to study the possibility of co-crystallizing the fluorene molecule, which presents a
C2v symmetry, with an electron withdrawing molecular unit of higher symmetry, in order to obtain a charge transfer (CT) complex. We have chosen the fluorene molecule because it is well known for its optical responses and, for these properties, its use is investigated in active optically designed materials [
1,
2,
3]. We employed, as fluorene counterparts, tetracyanoethylene (TCNE in
Figure 1), 1,2,4,5 tetracyanobenzene (TCNB) and 7,7,8,8-tetracyanoquinodimethane (TCNQ), all presenting
D2h symmetry. We were aware that the crystal structures of fluorene containing molecular complexes were disordered [
4,
5,
6,
7].
The three complexes have been characterized employing spectroscopic techniques (FTIR and Raman spectroscopy) and single crystal X-ray diffraction analysis. The analysis of the X-ray diffraction data was complicated by the difficulty in assigning the correct space group, because of the uncertainty on the presence or not of an inversion center and the possible disorder of the fluorene unit. Therefore some
ab initio periodic theoretical calculations have been performed, in order to solve the problem inherent to the interpretation of the crystal data. All the experimental results, together with the synthesis and the crystallization techniques, will be fully described in a separated paper [
8]. The aim of this paper is to show that sensible models of the crystal structures can be obtained by a two step procedure: (i) the disordered experimental structure is first separated into its components by graphical manipulation; (ii) then the geometry is optimized by means of
ab initio quantum-chemical calculations, carried out with the CRYSTAL code [
9]. The Hartree-Fock (HF) level of theory was adopted within the LCAO approximation and periodic boundary conditions. Recently, thanks to the implementation in CRYSTAL [
9] of the analytical HF nuclear gradient [
10,
11], geometry optimization
Figure 1.
Fluorene and its D2h counterpart in the co-crystalline complex (C-H bonds are indicated by dotted lines).
Figure 1.
Fluorene and its D2h counterpart in the co-crystalline complex (C-H bonds are indicated by dotted lines).
of periodic systems has become feasible in routine calculations [
12]. Thus crystal structures can be optimized and an
ab initio quantum mechanical structure refinement can be performed. In particular, here we report in details the above-described procedure, applied to the structure of the Fluorene/Tetracyanoethylene complex (hereafter named complex
1).
Results
From a Disordered Crystal Structure to an Ordered Model
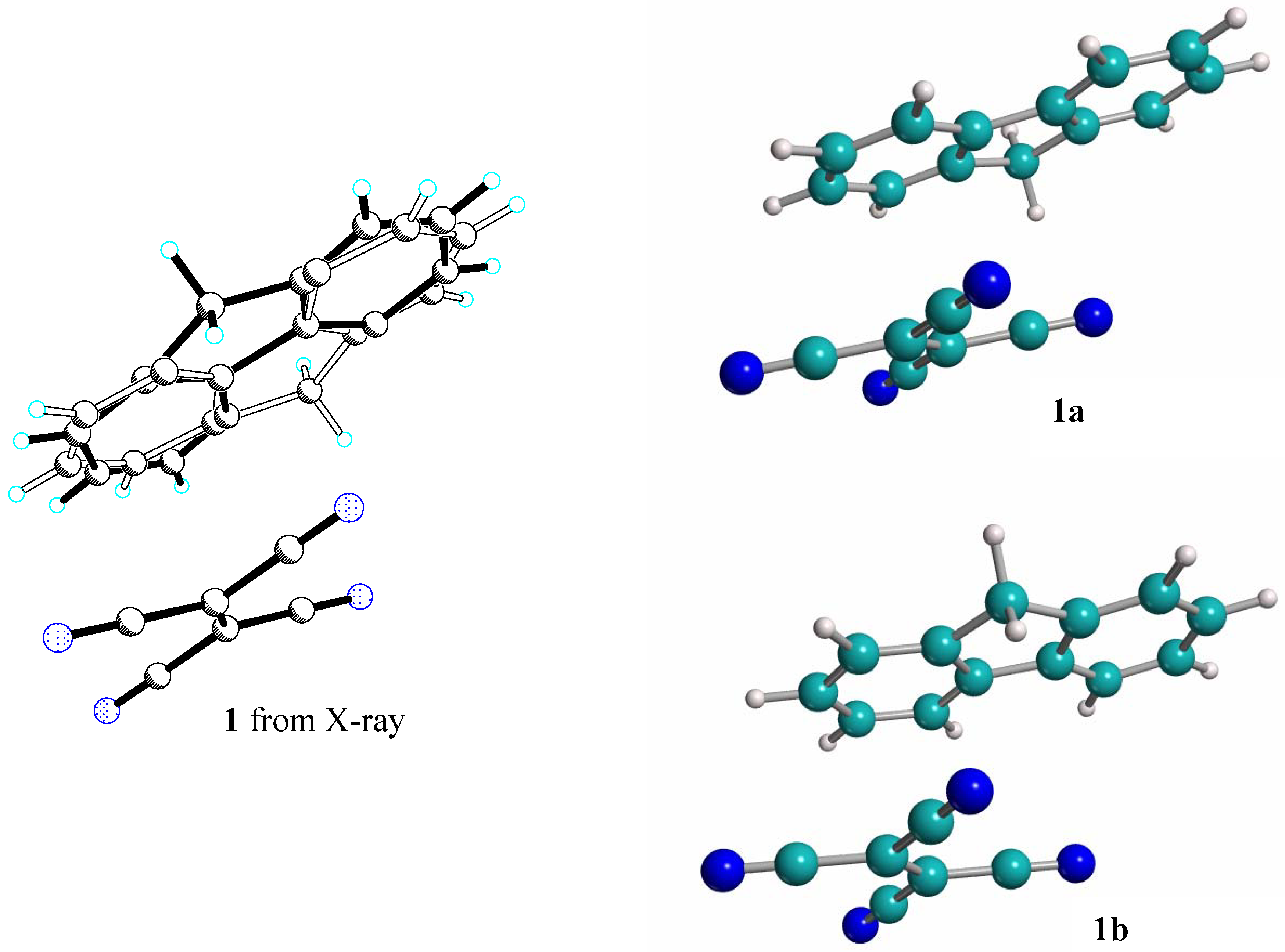
The crystal structure of complex
1 is disordered in the solid state and only an approximate structure can be obtained by refinement of the single crystal data. Fluorene (the electron donating C
2v molecule) always presents a very irregular geometry, showing for example C-C bonds ranging from 1.25Å to 1.75Å. Conversely, the electron withdrawing D
2h molecules (i.e. the counter part of fluorene in the complexes) are ordered, thanks to their higher symmetry as already described by Muhle et al. [
5]. Graphical inspection of the crystal structures suggests that the fluorene molecules in these co-crystalline complexes can assume two possible positions (indicated by black and white bonds in
Figure 2), related by an inversion center, both with 50% population.
Molecular manipulations by means of the MOLDRAW [
15] and XP [
16] programs were performed to generate the two possible models with the two ordered positions of the fluorene molecule without modifying the crystallographic periodicity. This artificial graphical separation process is illustrated in
Figure 2 for complex
1. The model crystal structure with the combination of the fluorene molecule
Figure 2.
Sketch of the graphical separation process used to obtain the two distinct ordered structures from the disordered crystal structure of 1, where the black and white bonds indicate the 1a and 1b positions respectively.
Figure 2.
Sketch of the graphical separation process used to obtain the two distinct ordered structures from the disordered crystal structure of 1, where the black and white bonds indicate the 1a and 1b positions respectively.
labeled
a with its TCNE counterpart is named
1a, and that with the combination of molecule
b with its TCNE counterpart is named
1b. Of course, the model ordered structures
1a and
1b, derived from the centrosymmetric P-1 X-ray structure, belong to the related non-centrosymmetric space group P1. Starting from these model structures their full geometry optimization was performed to obtain sensible geometries and to verify whether the
a and
b positions present the same stability. Besides, an estimate of the lattice energy (L.E.) was obtained by subtracting the gas phase energy of the isolated molecules from the crystal energy, according to eq. 1.
Geometry Optimization
In this paragraph we describe the geometric features of compounds
1a and
1b after geometry optimization of the model structures (
Figure 2) at the HF level, employing increasingly larger basis sets.
No significant changes in the values of bond distances and angles can be observed passing from the HF/3-21G to the HF/6-21G(d,p) level of theory and therefore only the results obtained at the HF/6-21G(d,p) level are reported.
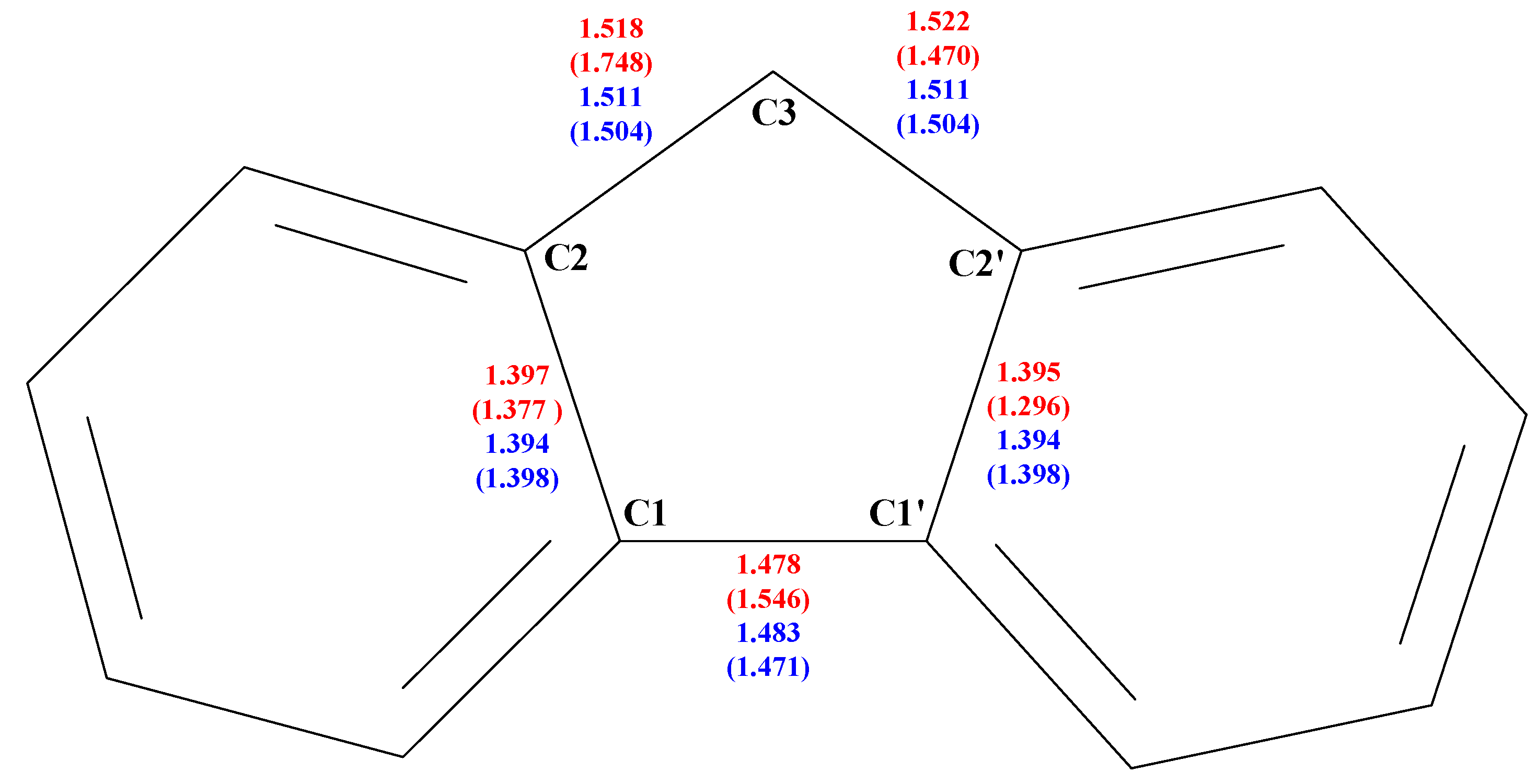
The more dramatic changes between the X-ray structure and that after geometry optimization involve the 5-member ring of fluorene, shown in
Figure 3 with the adopted labeling scheme.
Figure 3 and
Table 1 show the comparison between the geometrical features of the 5-member ring before and after full geometry optimization of
1a. It can be seen that the geometry optimization gives a chemically sensible model with the expected bond lengths and angles, starting from the rather deformed geometry obtained from the X-ray data. Indeed the optimized bond distances and angles (see
Figure 3 and
Table 1) are rather similar to those from the X-ray crystal structure of fluorene alone [
17], which is not disordered. The comparison between the geometry of fluorene alone in the crystal structure [
17] and of fluorene after HF/6-21G(d,p) full geometry optimization suggests that this level of theory is sufficiently accurate to describe the geometry of fluorene itself. Therefore the discrepancies between the calculated and experimental geometries of fluorene in complex
1 are certainly due to the structural disorder of the X-ray structure of complex
1.
Figure 3.
The 5-member ring of fluorene and its labeling. The values after geometry optimization are compared with the corresponding values from the X-ray data refinements (in parenthesis) of
1a (
in red) and of fluorene alone [
17] (
in blue).
Figure 3.
The 5-member ring of fluorene and its labeling. The values after geometry optimization are compared with the corresponding values from the X-ray data refinements (in parenthesis) of
1a (
in red) and of fluorene alone [
17] (
in blue).
Table 1.
Bond angles of the 5-member ring of fluorene in
1a fluorene alone [
17] from the X-ray data and after full geometry optimisation.
Table 1.
Bond angles of the 5-member ring of fluorene in 1a fluorene alone [17] from the X-ray data and after full geometry optimisation.
| Angle | 1a | 1a | Fluorene($) | Fluorene(*) |
| [°] | 6-21G(d,p) | X-ray | 6-21G(d,p) | X-ray |
| C1-C2-C3 | 110.3 | 102.4 | 110.4 | 110.4 |
| C2-C3-C2’ | 102.2 | 120.0 | 102.5 | 102.7 |
| C3-C2’-C1’ | 110.5 | 102.3 | 110.5 | 110.4 |
| C2’-C1’-C1 | 108.4 | 107.2 | 108.3 | 108.3 |
| C1’-C1-C2 | 108.5 | 128.3 | 108.7 | 108.3 |
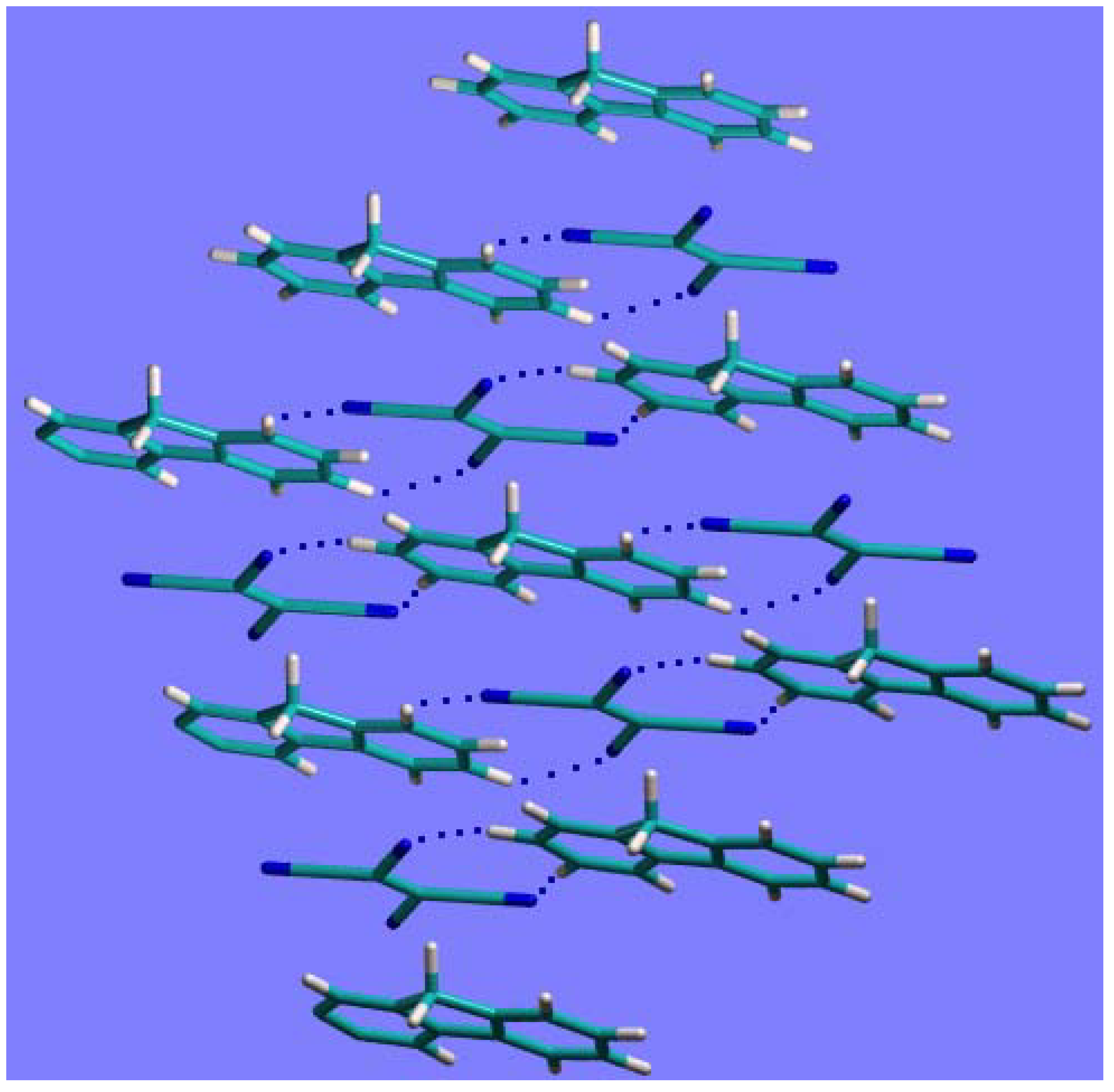
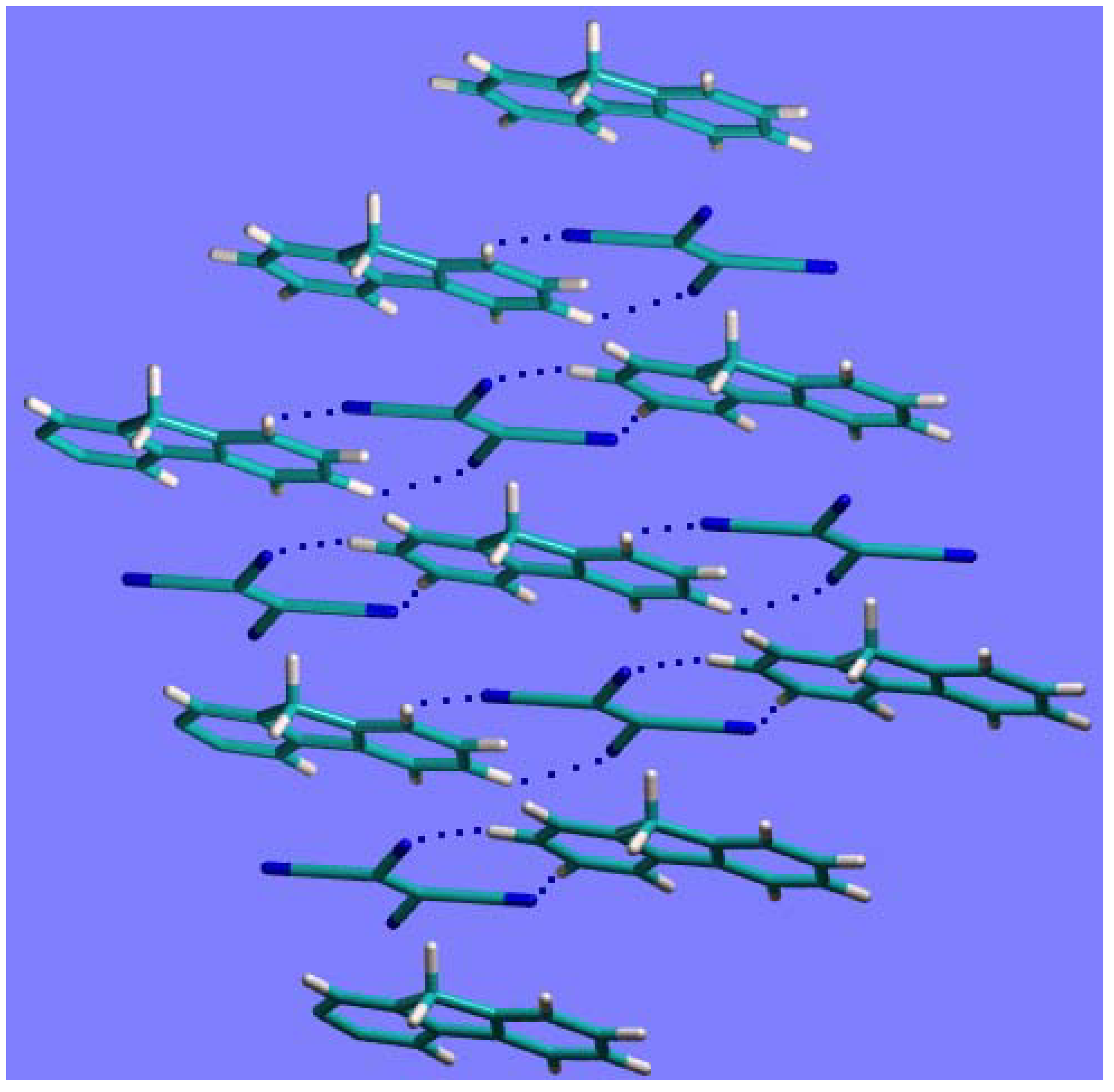
In
Figure 4 the crystal packing of the complex
1a, after geometry optimization at the HF/6-21G(d,p) level, is shown. The intermolecular interactions driving the crystal packing are:
the weak C-H·····N interactions between molecules on the same plane (dotted lines in
Figure 4), with H·····N distances in the range 2.55-2.91 Å;
the stacking interactions between the fluorene and TCNE molecules on adjacent parallel planes. The distance between the centers of the C=C bond in TCNE and of the facing phenyl group in fluorene (the two moieties, involved in the CT, shown in
Figure 4), is about 3.31Å. This value is very close to the inter-planar distance (~3.28Å), indicating an almost perfect facing between TCNE and the phenyl group in fluorene.
It is worth noting that the intermolecular distances obtained from the disordered crystal structures were too crude to allow a reliable discussion of the above packing geometry.
Figure 4.
The crystal packing arrangement of compound 1a after geometry optimization.
Figure 4.
The crystal packing arrangement of compound 1a after geometry optimization.
Energy Results
In
Table 2 the energetic features of the studied models after full geometry optimization are reported. The optimized structures
1a and
1b retain an enantiomeric relation and have very similar total energy values after geometry optimization. Indeed the two fluorene dispositions differ by less than 0.2 kJ/mol at the HF/6-21G(d,p) level of theory and therefore can be considered equivalent. The presence of the inversion center (relating the two equivalent fluorene positions in the actual disordered crystal structure) is then explained and confirmed.
As already mentioned, the described procedure also allows an approximate estimate of the lattice energy (see eq. 1). For instance the value calculated for
1 at the HF/3-21G level is: L.E. =136.2 kJ/mol. The comparison of the values obtained for different complexes can be correlated with the experimental spectroscopic evidences on the weak interactions holding together the complexes in the solid state and in solution (a detailed comparison will be reported in a separated paper [
8]).
Table 2.
Total energy for the two model structures of compound 1 (data in Hartrees, 1 Hartree = 2625.5 kJ/mol) and relative stability ΔE = E1a-E1b (in kJ/mol) at different levels of calculation.
Table 2.
Total energy for the two model structures of compound 1 (data in Hartrees, 1 Hartree = 2625.5 kJ/mol) and relative stability ΔE = E1a-E1b (in kJ/mol) at different levels of calculation.
| | 1a | 1b | ΔE |
| HF/3-21G | -937.7946535 | -937.7941936 | -1.21 |
| HF/6-21G | -942.0021077 | -942.0021458 | 0.10 |
| HF/6-21G(d) | -942.4568083 | -942.45694895 | 0.37 |
| HF/6-21G(d,p) | -942.4857835 | -942.4858601 | 0.20 |
Conclusions
In this work the CRYSTAL code was employed to elucidate the disordered crystal structure, obtained from X-ray single crystal analysis on a molecular complex of fluorene with the electron withdrawing TCNE molecule (
Figure 1). The crystal structure analysis of complex
1 (
Figure 2 left) indicated that the fluorene molecule is disordered over two symmetry related positions (
1a and
1b in
Figure 2 right) in the centrosymmetric
P-1 crystal structure.
Ab initio periodic calculations on the two graphically separated non-centrosymmetric
P1 model crystal structures indicated that these two positions are equivalent from the geometric and the energetic point of view. The presence of an inversion center is thus explained and confirmed and the correctness of the X-ray data refinement in the
P-1 space group is verified. The geometry of the disordered fluorene obtained in this way is poorly defined and only by the
ab initio full geometry optimization of the
P1 model structures could a sensible fluorene moiety, with the expected bond distances (
Figure 3) and angles (
Table 1) and intermolecular contacts (
Figure 4), be obtained, together with an estimate of the lattice energy.
The main scope of the present paper is to show that the deformed geometry obtained from the X-ray analysis of a disordered crystal structure can be improved by the use of the new features of the CRYSTAL code, which allow the
ab initio full geometry optimization of a periodic structure. A full account of the crystallographic and theoretical implications of the described procedure will be given elsewhere. [
18].




{kind=link}
{kind=link}
{kind=link}
{kind=link}