Effects of Xenoestrogens on T Lymphocytes: Modulation of bcl-2, p53, and Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Reagents and Cell Culture
2.2. Lymphoproliferation
2.3. Cell Cycle Analysis
2.4. DNA Fragmentation
2.5. Western blot analysis
2.6. RNA preparation and analysis
2.7. Statistical analysis
3. Results
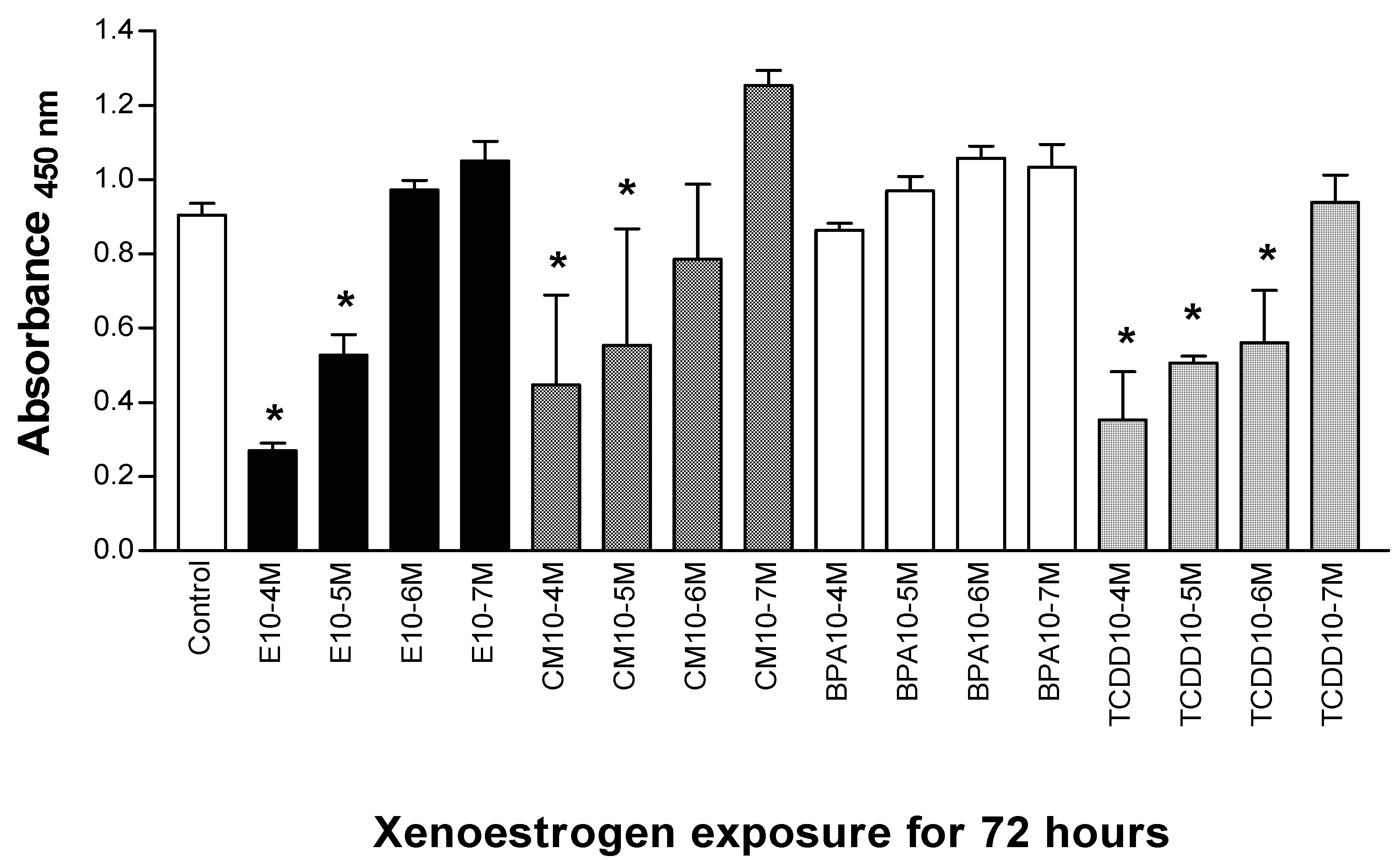
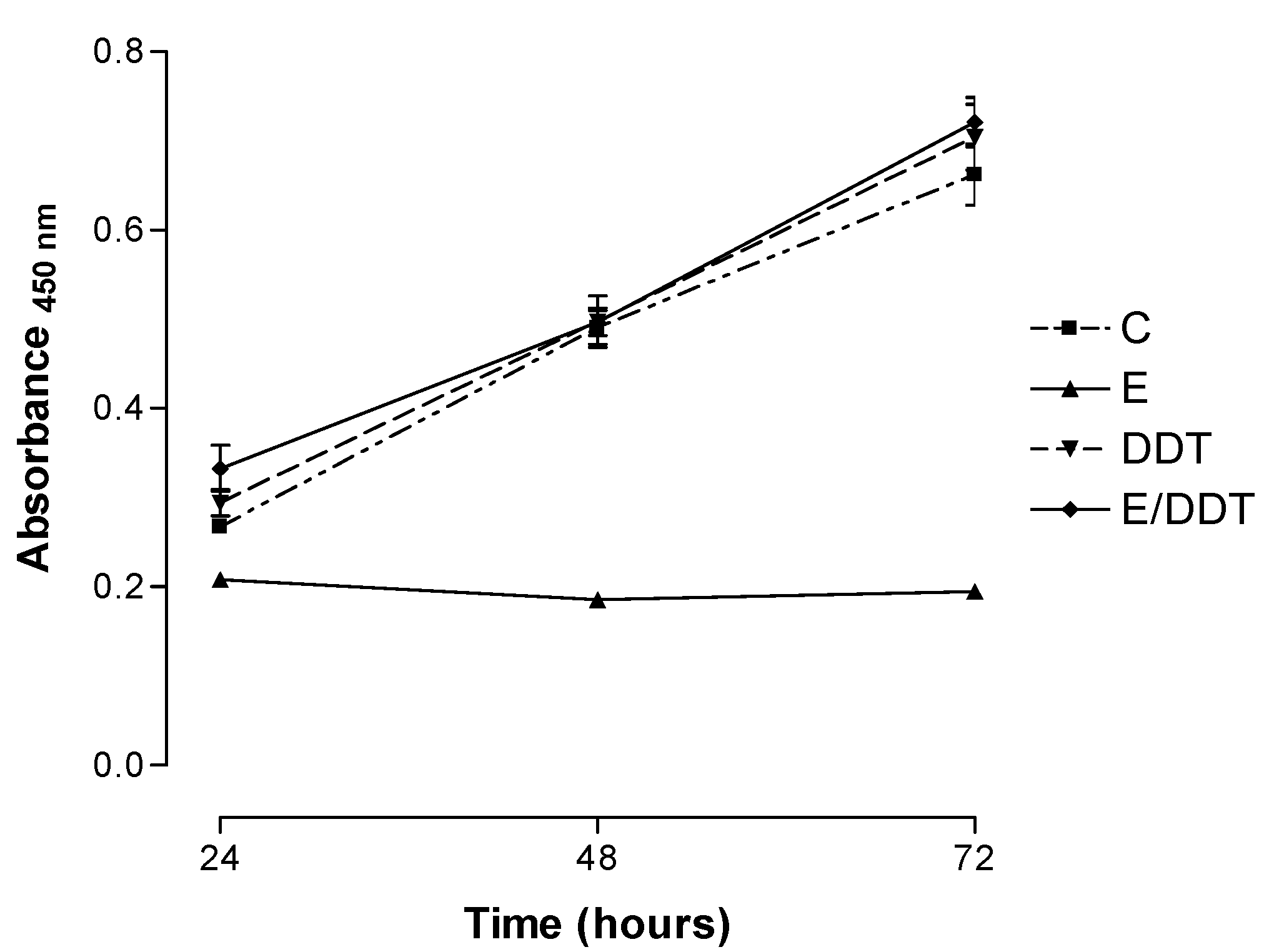
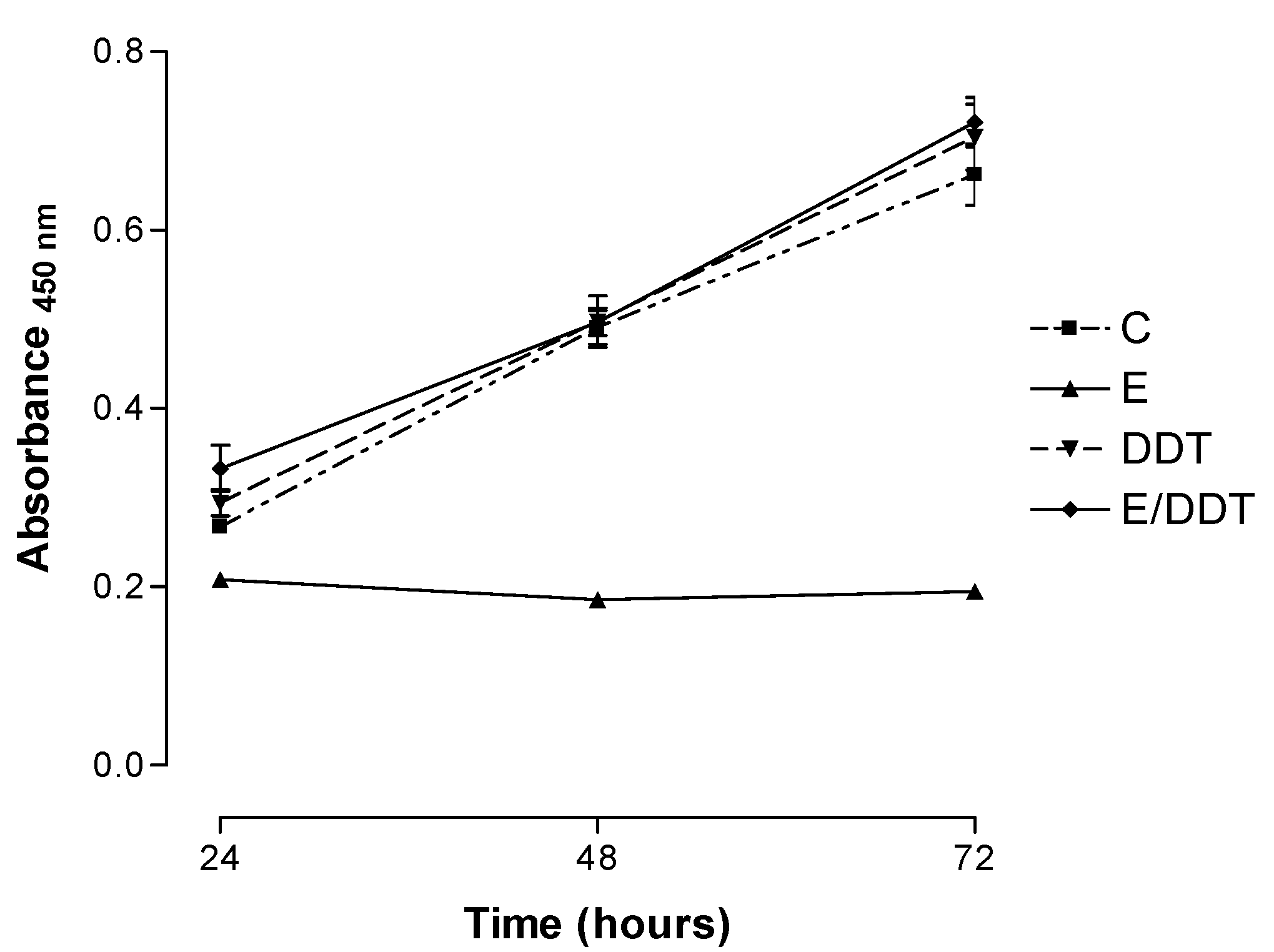
3.1. Xenoestrogen effects on Jurkat T cell proliferation
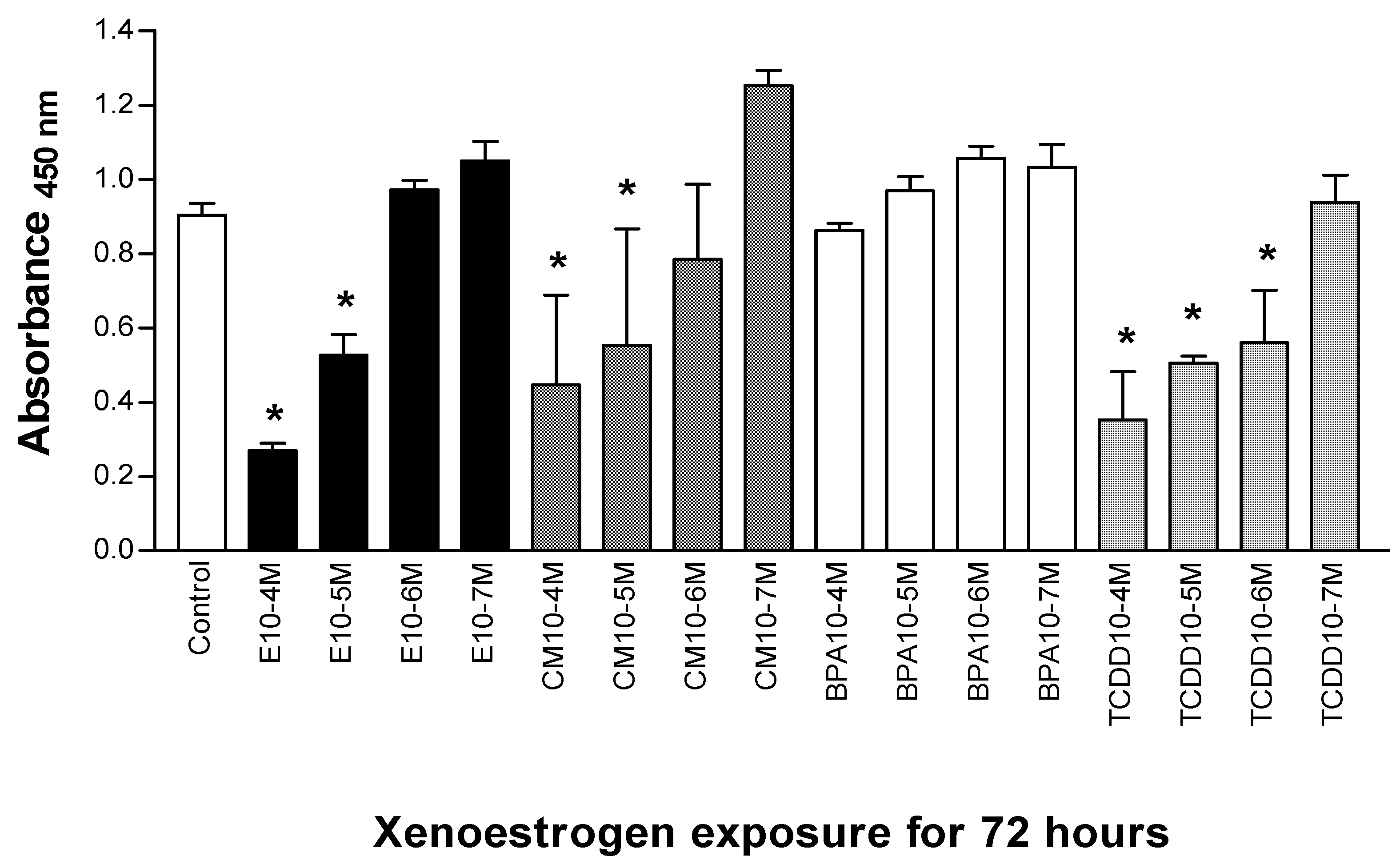
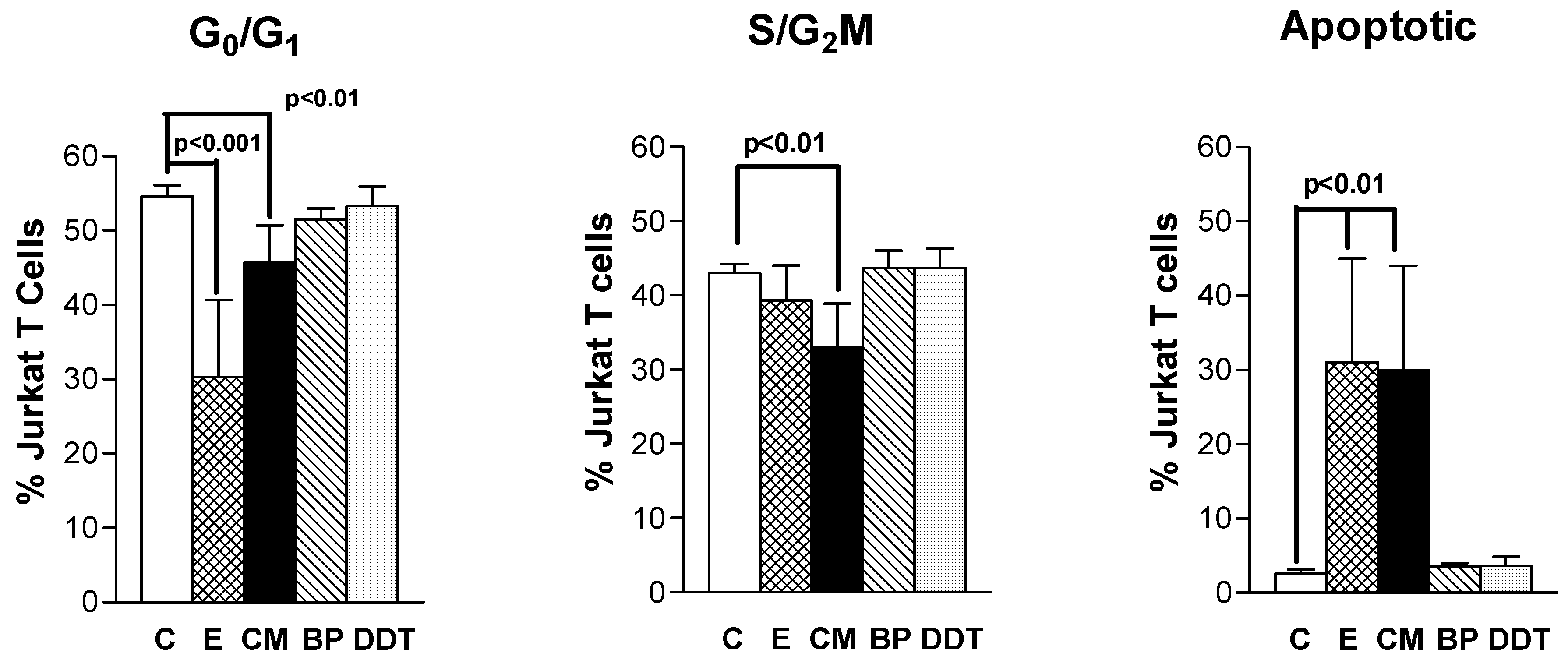
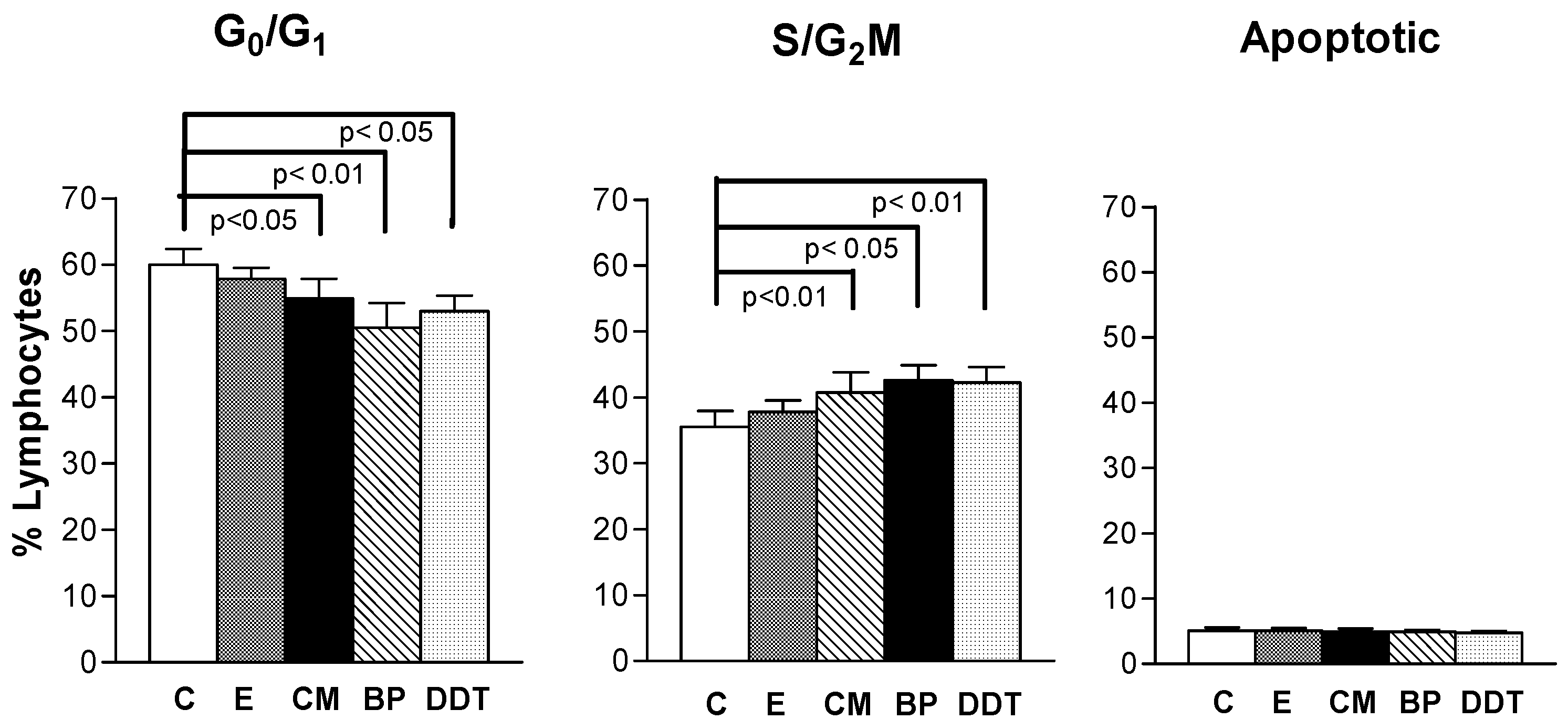
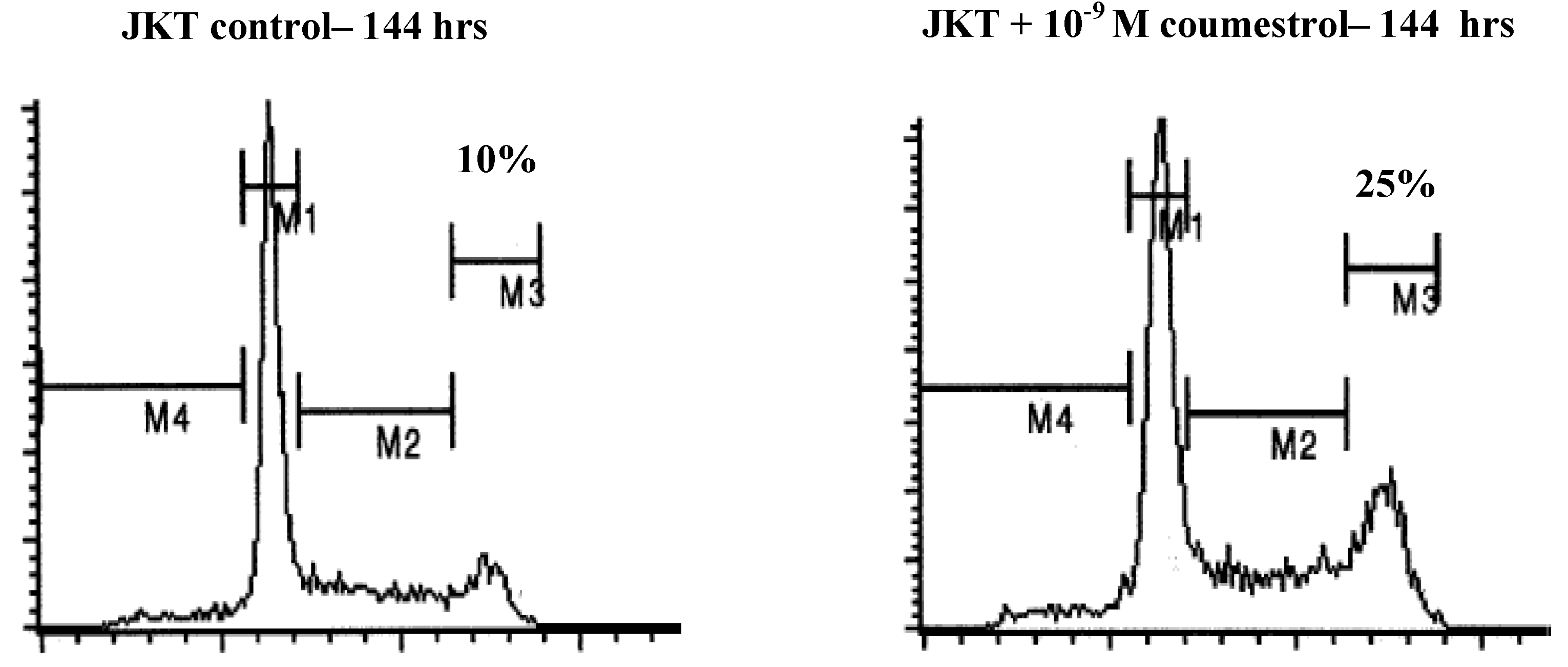
3.2. Xenoestrogen modulation of cell cycle phase distribution
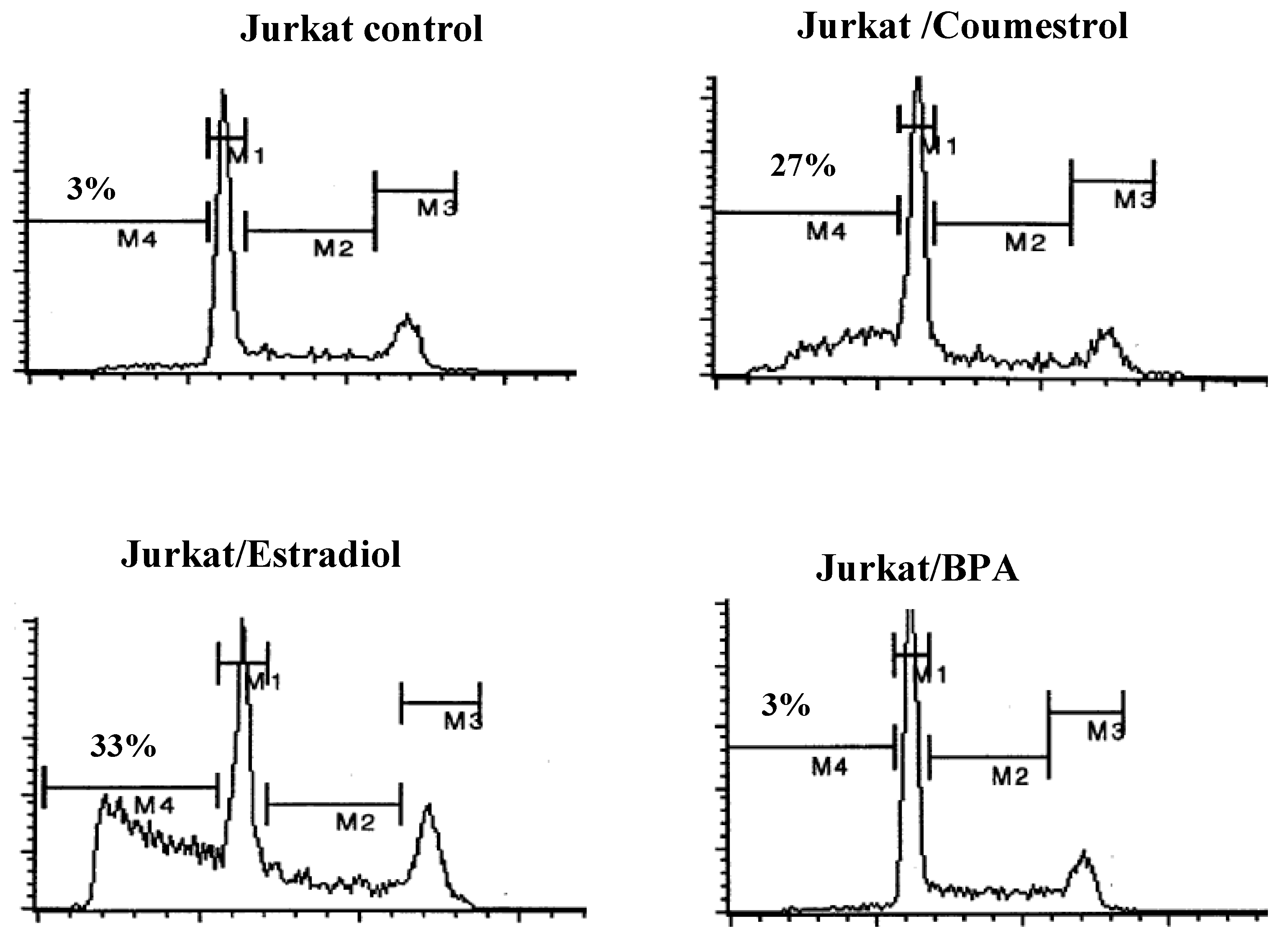
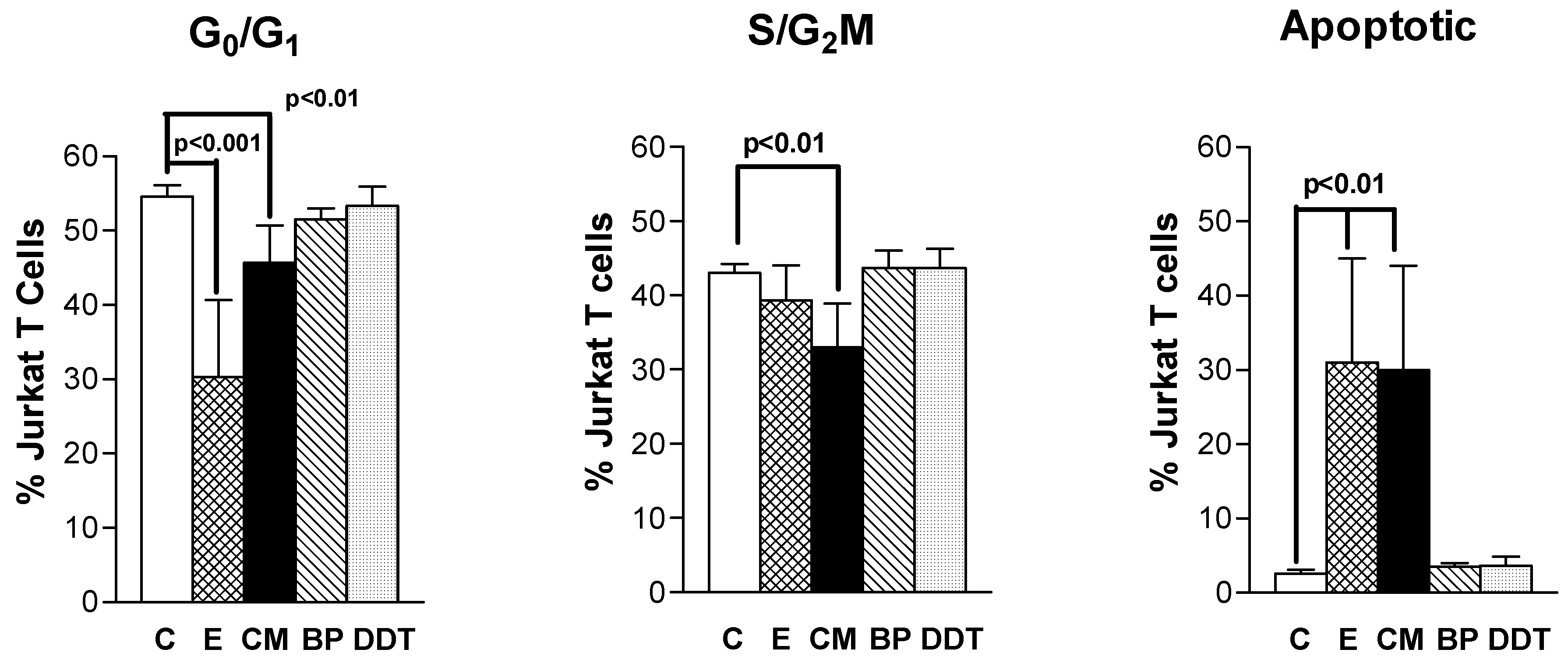
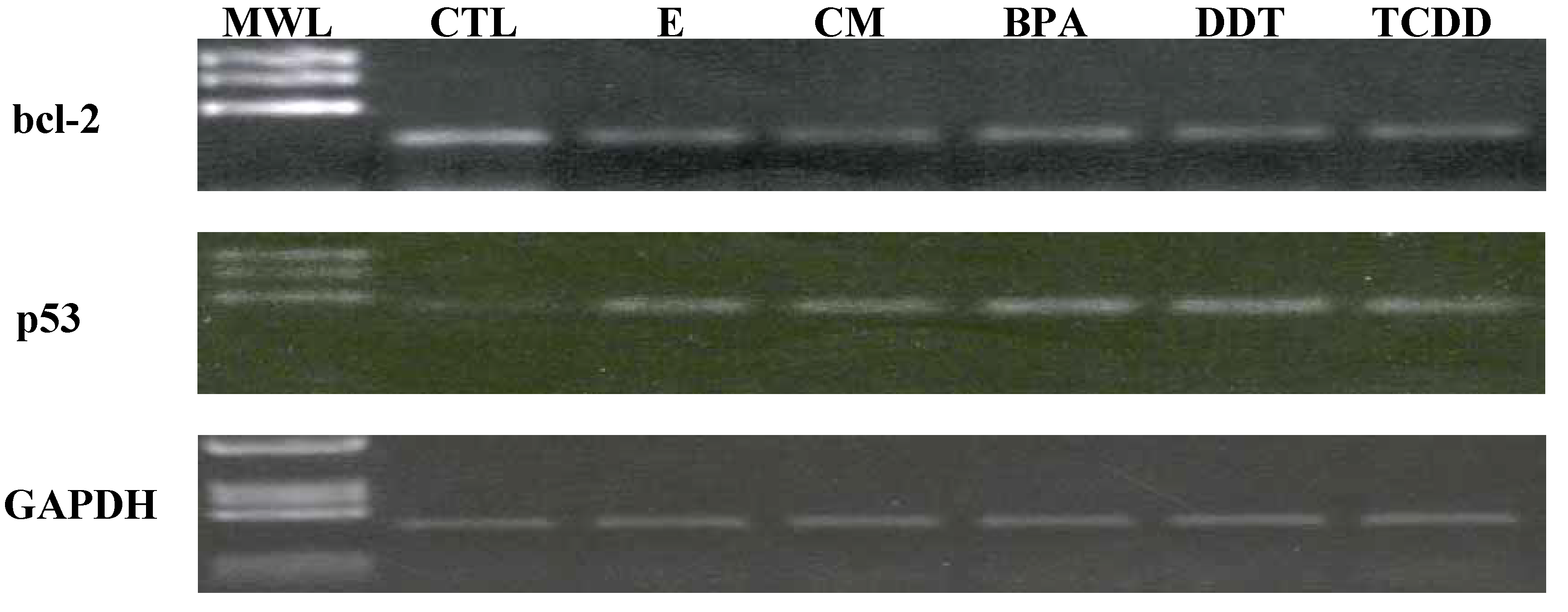
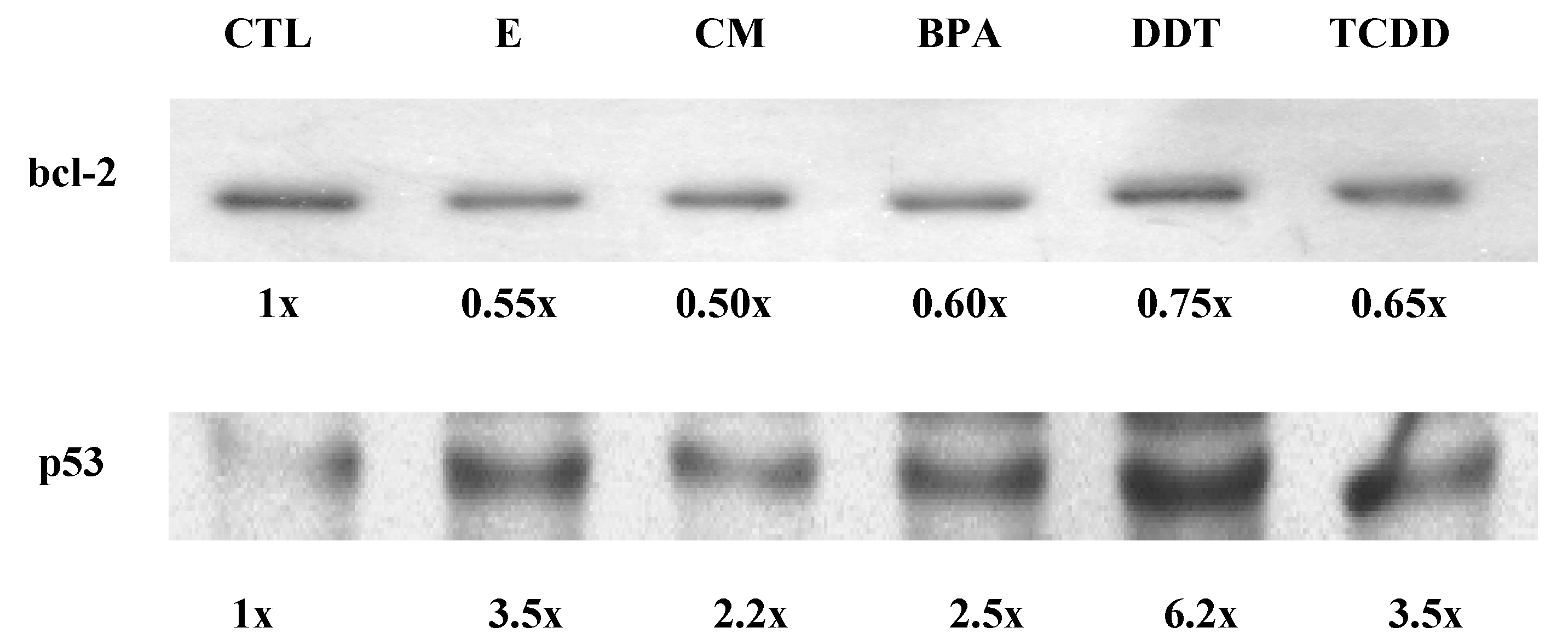
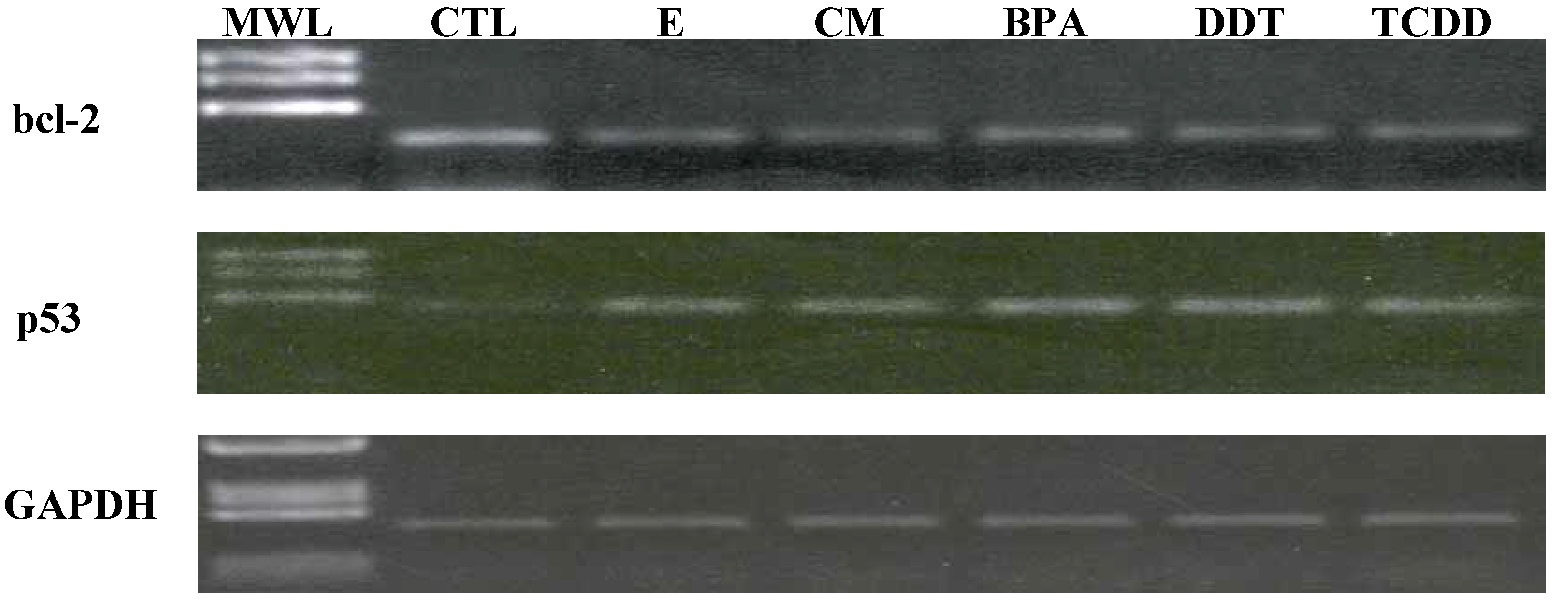
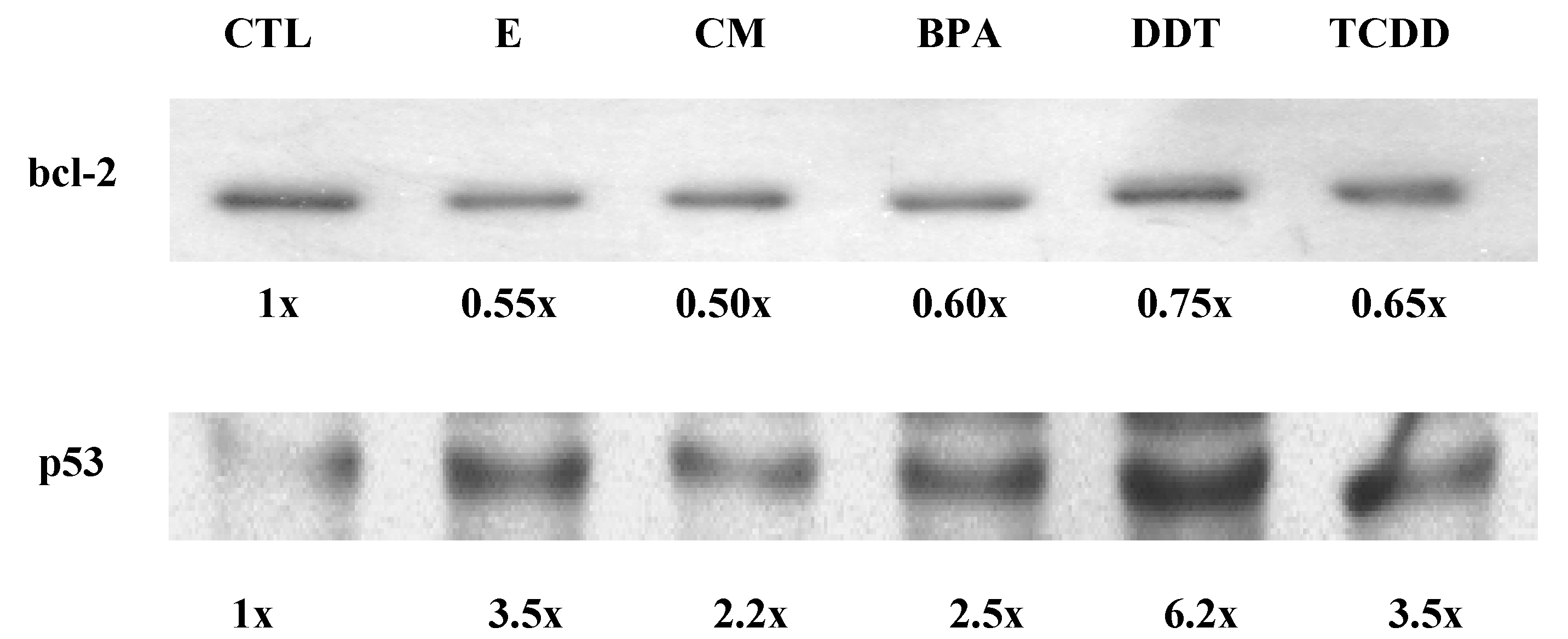
3.3. Xenoestrogen suppression of bcl-2 and stimulation of p53
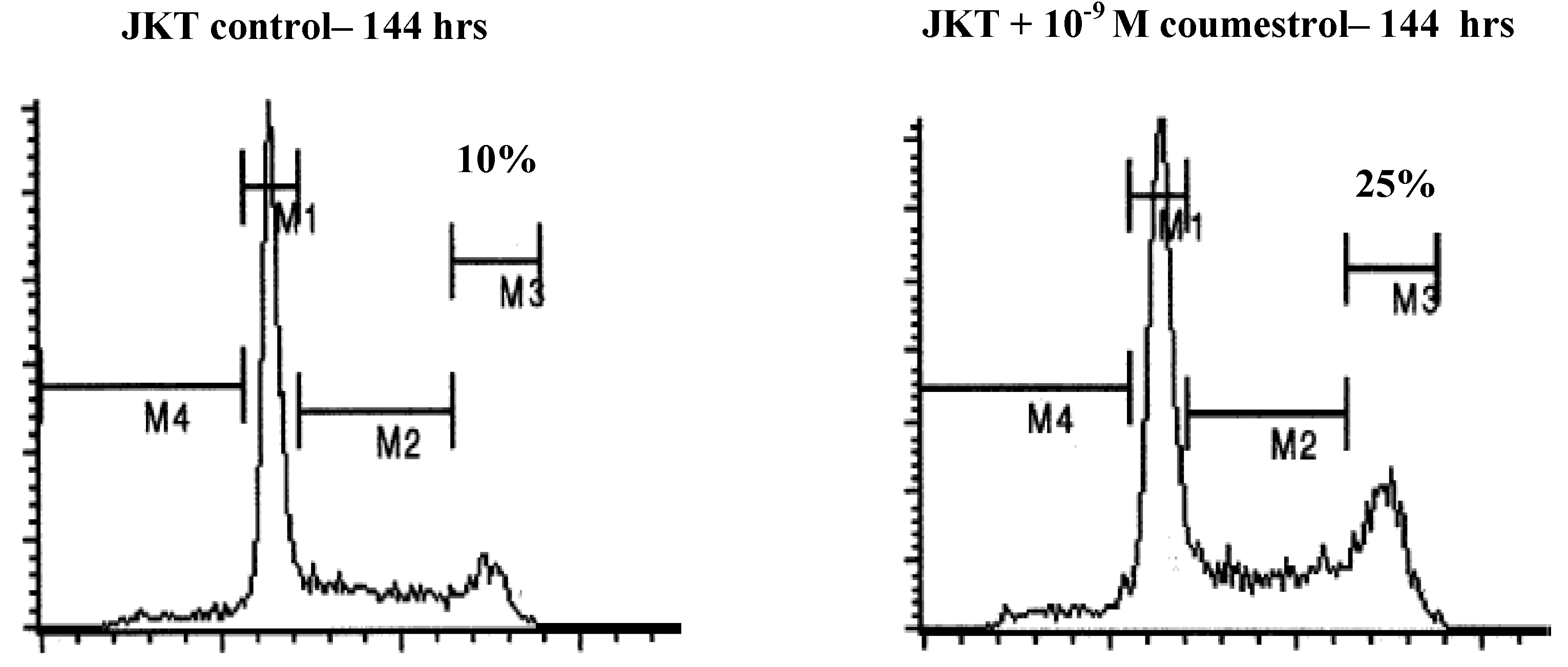
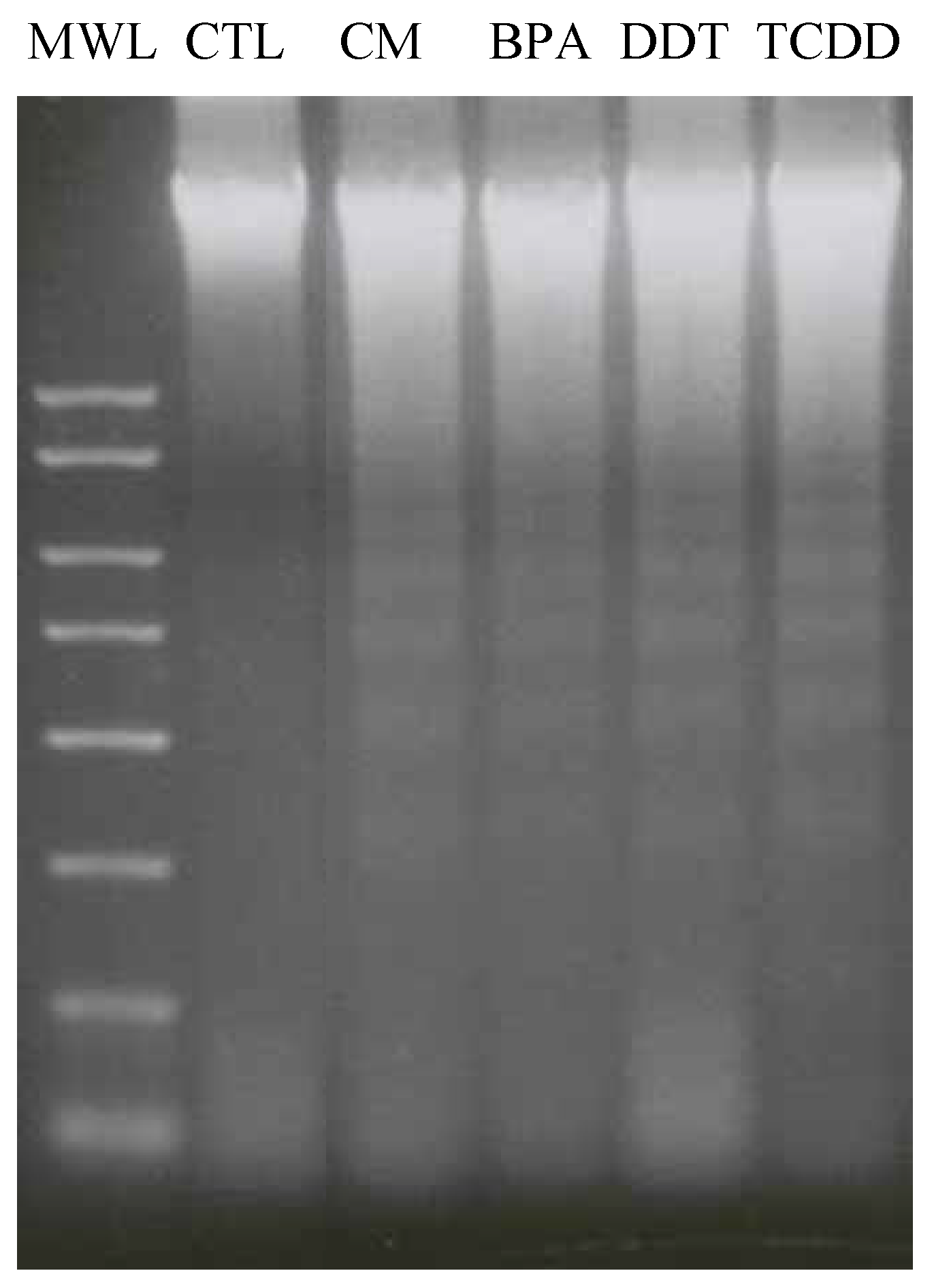
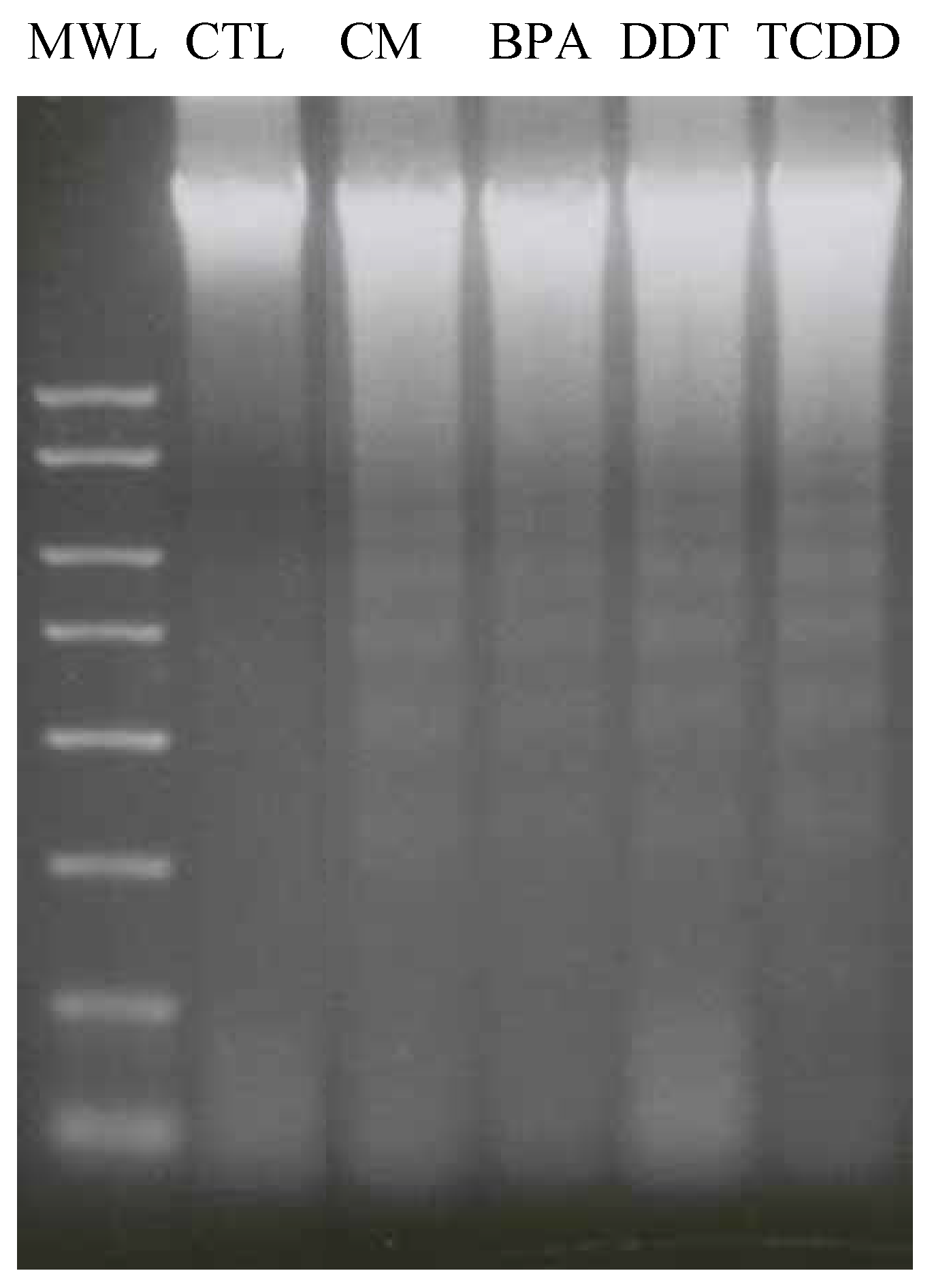
3.4. Xenoestrogen effects on apoptosis
3.5. Xenoestrogen effects on peripheral blood mononuclear cells

4. Discussion
Acknowledgements
References
- Whitacre, C.C.; Reingold, S.C.; O’Looney, P.A. Task Force on Gender, Multiple Sclerosis and Autoimmunity. A Gender Gap in Autoimmunity. Science 1999, 283, 1277–1278. [Google Scholar] and supplementary material at www.sciencemag.org/feature/data/983519.shl.
- Verthelyi, D. Sex hormones as immunomodulators in health and disease. Int. Immunopharmacol. 2001, 1, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.J.; Kovacs, W.J. Gonadal steroids and immunity. Endocrine. Rev. 1996, 17, 369–384. [Google Scholar]
- Fox, H.S. Sex steroids and the immune system. Ciba Foundation Symposium 1995, 191, 203–211. [Google Scholar] [PubMed]
- McMurray, R.W. Estrogen, prolactin, and autoimmunity: actions and interactions. Int. Immunopharmacol. 2001, 1, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.S. Environmental estrogens. Environ. Health Perspect. 1995, 103(9), 784–785. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.H. Endocrine disrupters and human health - is there a problem? An update. Environ. Health Perspect. 2000, 108(6), 487–93. [Google Scholar] [PubMed]
- Kaiser, J. Endocrine disrupters. Panel cautiously confirms low-dose effects. Science 2000, 290(5492), 695–697. [Google Scholar] [CrossRef] [PubMed]
- Neubert, D. Vulnerability of the endocrine system to xenobiotic influence. Reg. Toxicol. Pharmacol. 1997, 26, 9–29. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Hissong, B.D.; Verthelyi, D.; Donner, K.; Becker, K.; Karpuzoglu-Sahin, E. Gender and risk of autoimmune diseases: possible role of estrogenic compounds. Environ. Health Perspect. 1999, 5, 681–686. [Google Scholar] [CrossRef]
- Ashby, J. Testing for endocrine disruption post-EDSTAC: extrapolation of low dose rodent effects to humans. Toxicol. Lett. 2001, 120(1-3), 233–242. [Google Scholar] [PubMed]
- Crinnion, W.J. Environmental medicine, part one: the human burden of environmental toxins and their common health effects. Altern. Med. Rev. 2000, 5(1), 52–63. [Google Scholar] [PubMed]
- Ziegler, J. Environmental "endocrine disrupters" get a global look. J. Natl. Cancer Inst. 1997, 89(16), 1184–1187. [Google Scholar] [PubMed]
- Barton, H.A.; Andersen, M.E. Endocrine active compounds: from biology to dose response assessment. Crit. Rev. Toxicol. 1998, 28(4), 363–423. [Google Scholar] [CrossRef] [PubMed]
- Barton, H.A.; Andersen, M.E. Dose-response assessment strategies for endocrine-active compounds. Regul. Toxicol. Pharmacol. 1997, 25(3), 292–305. [Google Scholar] [CrossRef] [PubMed]
- Domon, O.E.; McGarrity, L.J.; Bishop, M.; Yoshioka, M.; Chen, J.J.; Morris, S.M. Evaluation of the genotoxicity of the phytoestrogen, coumestrol, in AHH-1 TK(+/-) human lymphoblastoid cells. Mutat. Res. 2001, 474(1-2), 129–37. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, K.; Okuma, M.; Karaki, S.; Matsuura, S.; Yoshida, T.; Aikawa, H.; Izumi, S.; Kayama, F. Inhibitory effect of natural and environmental estrogens on thymic hormone production in thymus epithelial cell culture. Int. J. Immunopharmacol. 1999, 21(12), 861–868. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, H.; Tsutsumi, O.; Momoeda, M.; Takai, Y.; Osuga, Y.; Taketani, Y. Differential interactions of bisphenol A and 17beta-estradiol with estrogen receptor alpha (ERalpha) and ERbeta. Endocr. J. 1999, 46(6), 773–778. [Google Scholar] [CrossRef] [PubMed]
- Howdeshell, K.L.; Hotchkiss, A.K.; Thayer, K.A.; Vandenbergh, J.G.; vom Saal, F.S. Exposure to bisphenol A advances puberty. Nature 1999, 401(6755), 763–764. [Google Scholar] [CrossRef] [PubMed]
- Sakazaki, H.; Ueno, H.; Nakamuro, K. Estrogen receptor alpha in mouse splenic lymphocytes: possible involvement in immunity. Toxicol. Lett. 2002, 133(2-3), 221–229. [Google Scholar] [PubMed]
- Tapiero, H.; Ba, G.N.; Tew, K.D. Estrogens and environmental estrogens. Biomed. Pharmacother. 2002, 56(1), 36–44. [Google Scholar] [CrossRef] [PubMed]
- Street, J.C.; Sharma, R.P. Alteration of induced cellular and humoral immune responses by pesticides and chemicals of environmental concern: quantitative studies of immunosuppression by DDT, aroclor 1254, carbaryl, carbofuran, and methylparathion. Toxicol. Appl. Pharmacol. 1975, 32(3), 587–602. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.D.; Koner, B.C.; Ray, A. Influence of stress on DDT-induced humoral immune responsiveness in mice. Environ. Res. 1997, 74(1), 43–7. [Google Scholar] [CrossRef] [PubMed]
- Koner, B.C.; Banerjee, B.D.; Ray, A. Organochlorine pesticide-induced oxidative stress and immune suppression in rats. Indian J. Exp. Biol. 1998, 36(4), 395–8. [Google Scholar] [PubMed]
- Dees, C.; Askari, M.; Foster, J.S.; Ahamed, S.; Wimalasena, J. DDT mimicks estradiol stimulation of breast cancer cells to enter the cell cycle. Mol. Carcinog. 1997, 18(2), 107–114. [Google Scholar] [CrossRef] [PubMed]
- Diel, P.; Olff, S.; Schmidt, S.; Michna, H. Effects of the environmental estrogens bisphenol A, o,p′-DDT, p-tert-octylphenol and coumestrol on apoptosis induction, cell proliferation and the expression of estrogen sensitive molecular parameters in the human breast cancer cell line MCF-7. J. Steroid. Biochem. Mol. Biol. 2002, 80(1), 61–70. [Google Scholar] [CrossRef] [PubMed]
- Tebourbi, O.; Rhouma, K.B.; Sakly, M. DDT induces apoptosis in rat thymocytes. Bull. Environ. Contam. Toxicol. 1998, 61(2), 216–223. [Google Scholar] [CrossRef] [PubMed]
- Rininger, J.A.; Stoffregen, D.A.; Babish, J.G. Murine hepatic p53, RB, and CDK inhibitory protein expression following acute 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure. Chemosphere 1997, 34(5-7), 1557–68. [Google Scholar] [PubMed]
- Silverstone, A.E.; Frazier, D.E., Jr.; Fiore, N.C.; Soults, J.A.; Gasiewicz, T.A. Dexamethasone, beta-estradiol, and 2,3,7,8-tetrachlorodibenzo-p-dioxin elicit thymic atrophy through different cellular targets. Toxicol. Appl. Pharmacol. 1994, 126(2), 248–59. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.B.; Xu, H.; Nagarkatti, P.S.; Nagarkatti, M. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol. Appl. Pharmacol. 1997, 142(2), 367–77. [Google Scholar] [CrossRef] [PubMed]
- Prell, R.A.; Oughton, J.A.; Kerkvliet, N.I. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on anti-CD3-induced changes in T-cell subsets and cytokine production. Int. J. Immunopharmacol. 1995, 17(11), 951–961. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.W.; Hundeiker, C.; Gleichmann, E.; Esser, C. Cytokine gene expression during ontogeny in murine thymus on activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol. Pharmacol. 1997, 52(1), 30–37. [Google Scholar] [PubMed]
- Jeon, M.S.; Esser, C. The murine IL-2 promoter contains distal regulatory elements responsive to the Ah receptor, a member of the evolutionarily conserved bHLH-PAS transcription factor family. J. Immunol. 2000, 165(12), 6975–6983. [Google Scholar] [CrossRef] [PubMed]
- Kharat, I.; Saatcioglu, F. Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated by direct transcriptional interference with the liganded estrogen receptor. Cross-talk between aryl hydrocarbon- and estrogen-mediated signaling. J. Biol. Chem. 1996, 271(18), 10533–10537. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Tsuchiya, S.; Minegishi, M.; Osada, M.; Ikawa, S.; Tezuka, F.A.; Kaji, M.; Konno, T.; Watanabe, M.; Kikuchi, H. The Ah receptor is not involved in 2,3,7,8-tetrachlorodibenzo- p-dioxin-mediated apoptosis in human leukemic T cell lines. J. Biol. Chem. 1998, 273(31), 19853–1958. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Neckers, L.M. Cytostatic and cytotoxic activity of sex steroids against human leukemia cell lines. Cancer Letters 1994, 76, 81. [Google Scholar] [CrossRef] [PubMed]
- Kincade, P.W.; Medina, K.L.; Smithson, G. Sex hormones as negative regulators of lymphopoiesis. Immunol. Rev. 1994, 37, 119. [Google Scholar] [CrossRef]
- Jenkins, J.K.; Suwannaroj, S.; Elbourne, K.B.; Ndebele, K.; McMurray, R.W. 17-β-estradiol alters Jurkat lymphocyte cell cycling and induces apoptosis through suppression of bcl-2 and cyclin A. Internat. J. Immunopharmacol. 2001, 11, 1897–1911. [Google Scholar]
- McMurray, R.W.; Suwannaroj, S.; Ndebele, K.; Jenkins, J.K. Differential effects of sex steroids on T and B lymphocytes: modulation of cell cycling, apoptosis, and bcl-2. Pathobiol. 2001, 69, 44–58. [Google Scholar] [CrossRef]
- McMurray, R.W.; Ndebele, K.; Jenkins, J.K. 17-β-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991, 139, 271. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.J. Programmed cell death in the immune system. Adv. Immunol. 1991, 50, 55. [Google Scholar] [PubMed]
- King, K.L.; Cidlowski, J.A. Cell cycle and apoptosis: common pathways to life and death. J. Cell Biochem. 1995, 58, 175. [Google Scholar] [CrossRef] [PubMed]
- Pagliacci, M.C.; Spinozzi, F.; Migliorati, G.; Fumi, G.; Smacchia, M.; Grignani, F.; Riccardi, C.; Nicoletti, I. Genistein inhibits tumour cell growth in vitro but enhances mitochondrial reduction of tetrazolium salts: a further pitfall in the use of the MTT assay for evaluating cell growth and survival. Eur. J. Cancer 1993, 29A(11), 1573–1577. [Google Scholar] [CrossRef]
- Neumann, C.M.; Oughton, J.A.; Kerkvliet, N.I. Anti-CD3-induced T-cell activation--II. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Intenat. J. Immunopharmacol. 1993, 15(4), 543–550. [Google Scholar] [CrossRef]
- Huang, D.C.; O’Reilly, L.A.; Strasser, A.; Cory, S. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO Journal 1997, 16, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Holcombe, R.F.; Jain, S.K.; Alvarez-Hernandez, X.; Chervenak, R.; Wolf, R.E.; Glass, J. Evidence for the induction of apoptosis by endosulfan in a human T-cell leukemic line. Mol. Cell. Biochem. 2000, 205(1-2), 53–66. [Google Scholar] [PubMed]
- Roy, D.; Palangat, M.; Chen, C.W.; Thomas, R.D.; Colerangle, J.; Atkinson, A.; Yan, Z.J. Biochemical and molecular changes at the cellular level in response to exposure to environmental estrogen-like chemicals. J. Toxicol. Environ. Health 1997, 50(1), 1–29. [Google Scholar] [CrossRef] [PubMed]
- Rininger, J.A.; Stoffregen, D.A.; Babish, J.G. Murine hepatic p53, RB, and CDK inhibitory protein expression following acute 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure. Chemosphere 1997, 34(5-7), 1557–68. [Google Scholar] [PubMed]
- Burow, M.E.; Tang, Y.; Collins-Burow, B.M.; Krajewski, S.; Reed, J.C.; McLachlan, J.A.; Beckman, B.S. Effects of environmental estrogens on tumor necrosis factor alpha-mediated apoptosis in MCF-7 cells. Carcinogenesis 1999, 20(11), 2057–61. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.; Rajapakse, N.; Wilkins, M.; Kortenkamp, A. Prediciton and assessment of the effects of ixtures of four xenoestrogens. Env. Health Perspect. 2000, 108, 983–987. [Google Scholar] [CrossRef]
- Hughes, C.C.W.; Pober, J.S. Transcriptional regulation of the interleukin-2 gene in normal human peripheral blood T cells. J. Biol. Chem. 1996, 271, 5369–5377. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Wiskocil, R.L.; Stobo, J.D. The role of T3 surface molecules in the activation of human T cells: A two stimulus requirement for IL-2 production reflects events occurring at a pre-translational level. J. Immunol 1984, 133, 123–128. [Google Scholar]
- Landegren, U.; Andersson, J.; Wigzell, H. Analysis of human T lymphocyte activation in a T cell tumor model system. Eur. J. Immunol. 1985, 15, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Littman, D.R. Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.L.; Hamilton, J.A.; Sweeney, K.J.E.; Watts, C.K.; Musgrove, E.A. Steroidal regulation of the cell cycle. Ciba Foundation Symposium 1995, 191, 218. [Google Scholar] [PubMed]
- McDonnell, D.P.; Dana, S.L.; Hoener, P.A.; Lieberman, B.A.; Imhof, M.O.; Stein, R.B. Cellular mechanisms which distinguish between hormone and antihormone activated estrogen receptor. Ann. New York Acad. Sci. 1995, 761, 121–137. [Google Scholar] [CrossRef]
- Staples, J.E.; Fiore, N.C.; Frazier, D.E., Jr.; Gasiewicz, T.A.; Silverstone, A.E. Overexpression of the anti-apoptotic oncogene, bcl-2, in the thymus does not prevent thymic atrophy induced by estradiol or 2,3,7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1998, 151(1), 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sohoni, P.; Sumpter, J.P. Several environmental oestrogens are also anti-androgens. J. Endocrinol. 1998, 158(3), 327–39. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Lieberman, M.E. Estrogen-stimulated prolactin synthesis in vitro. Classification of agonist, partial agonist, and antagonist actions based on structure. Mol. Pharmacol. 1984, 26(2), 279–85. [Google Scholar] [PubMed]
© 2003 by MDPI (http://www.mdpi.org).
Share and Cite
Ndebele, K.; Tchounwou, P.B.; McMurray, R.W. Effects of Xenoestrogens on T Lymphocytes: Modulation of bcl-2, p53, and Apoptosis. Int. J. Mol. Sci. 2003, 4, 45-61. https://doi.org/10.3390/i4020045
Ndebele K, Tchounwou PB, McMurray RW. Effects of Xenoestrogens on T Lymphocytes: Modulation of bcl-2, p53, and Apoptosis. International Journal of Molecular Sciences. 2003; 4(2):45-61. https://doi.org/10.3390/i4020045
Chicago/Turabian StyleNdebele, Kenneth, Paul B. Tchounwou, and Robert W. McMurray. 2003. "Effects of Xenoestrogens on T Lymphocytes: Modulation of bcl-2, p53, and Apoptosis" International Journal of Molecular Sciences 4, no. 2: 45-61. https://doi.org/10.3390/i4020045
APA StyleNdebele, K., Tchounwou, P. B., & McMurray, R. W. (2003). Effects of Xenoestrogens on T Lymphocytes: Modulation of bcl-2, p53, and Apoptosis. International Journal of Molecular Sciences, 4(2), 45-61. https://doi.org/10.3390/i4020045