Post Hartree–Fock and DFT Studies on Pyrrole···Nitrogen and Pyrrole···Carbon Monoxide Molecules

Abstract
:Introduction
Computational details
Results and discussion
Geometries and energies
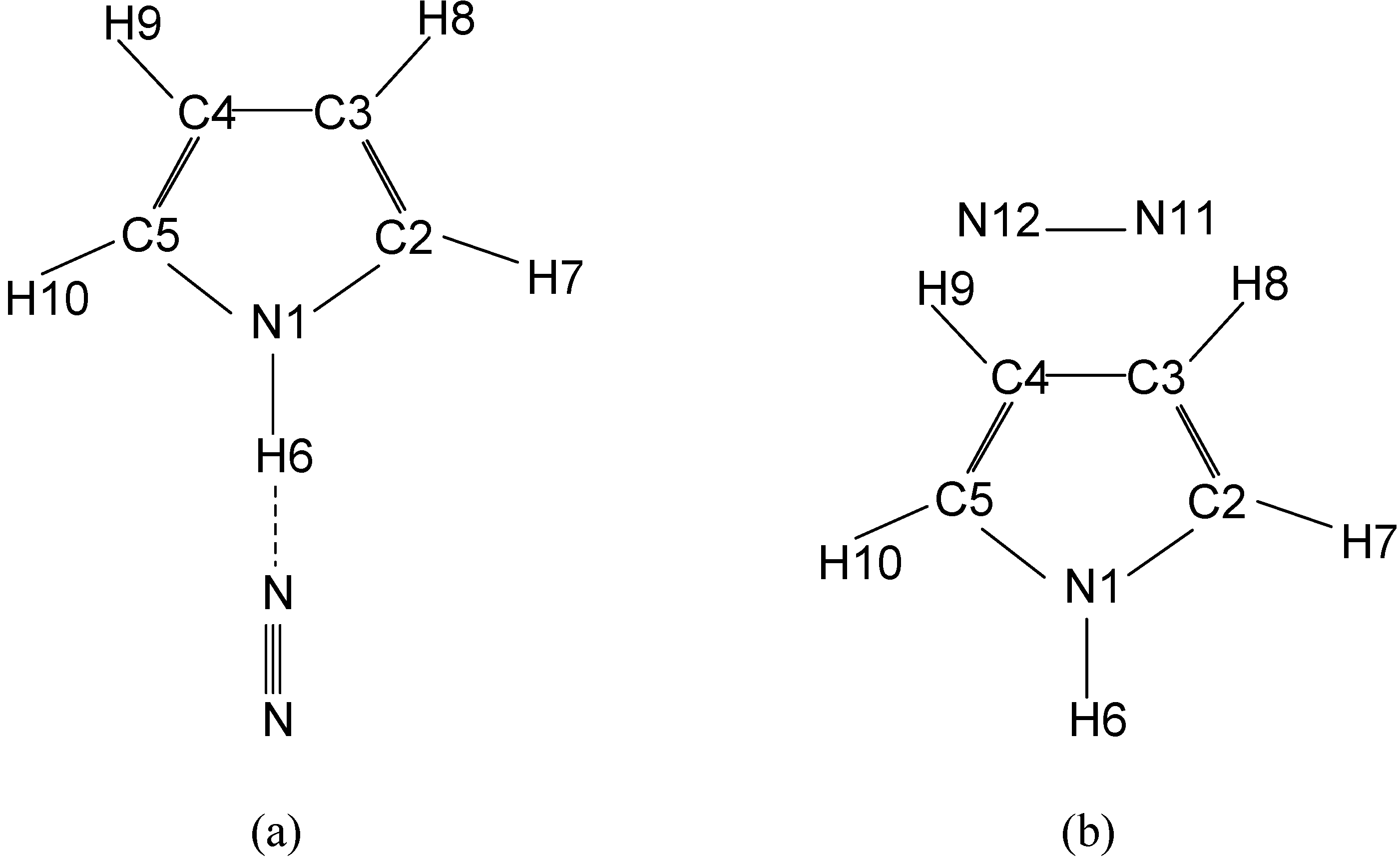

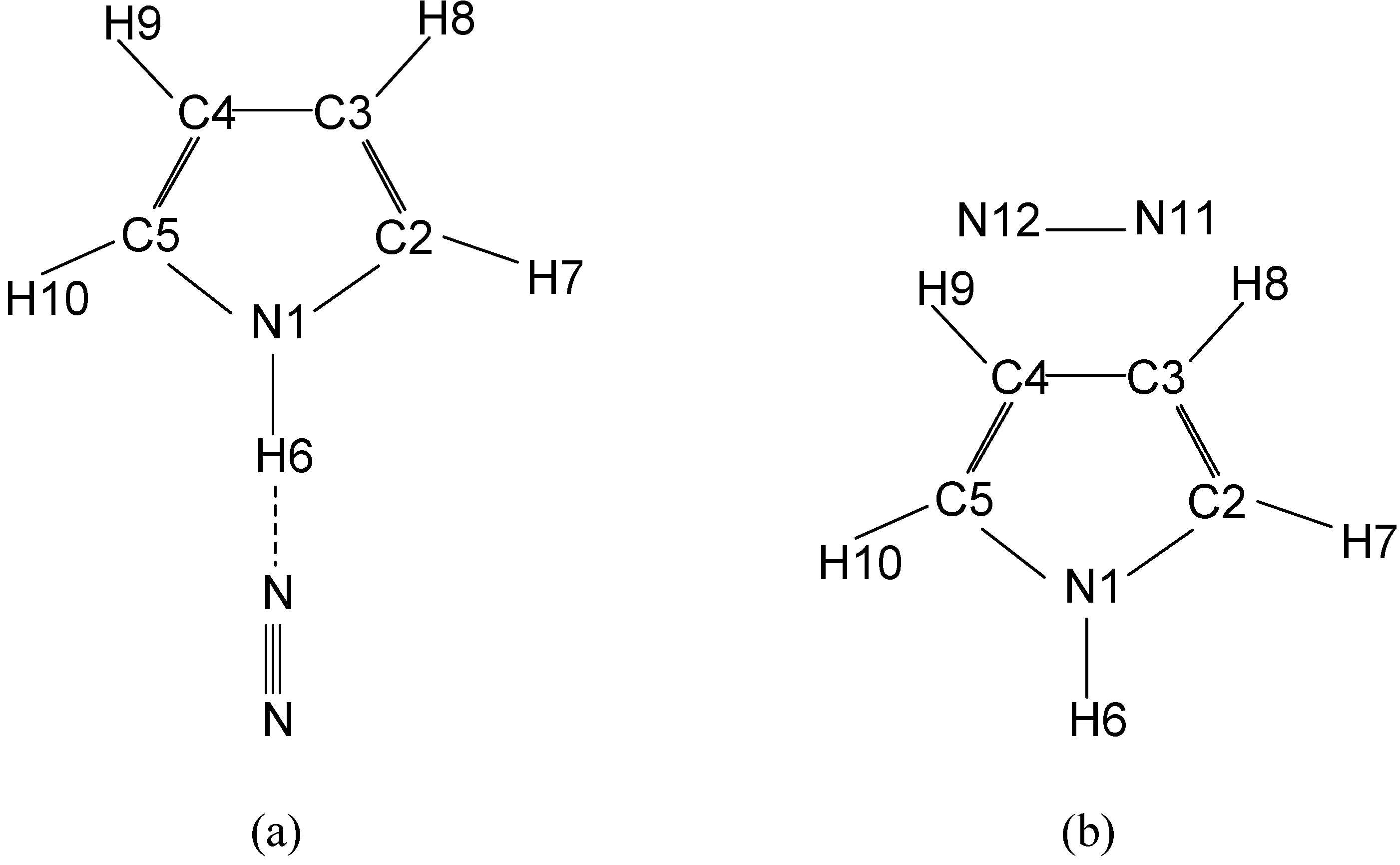{kind=link}
{kind=link}
| Parameters | Linear | T-shaped | Experimental value | |||||||||
| HF | MP2 | B3LYP | B3PW91 | HF | MP2 | B3LYP | B3PW91 | MW (Ref. 28) | MW corrected (Ref. 29) | |||
| R(N1-C2) | 1.362 | 1.373 | 1.375 | 1.370 | 1.363 | 1.373 | 1.376 | 1.371 | 1.370 | 1.370 | ||
| R(C2-C3) | 1.358 | 1.383 | 1.379 | 1.377 | 1.358 | 1.383 | 1.378 | 1.377 | 1.382 | 1.382 | ||
| R(C3-C4) | 1.426 | 1.418 | 1.425 | 1.422 | 1.427 | 1.419 | 1.426 | 1.422 | 1.417 | 1.417 | ||
| R(N1-H6) | 0.993 | 1.012 | 1.009 | 1.008 | 0.992 | 1.011 | 1.008 | 1.007 | 0.996 | 1.000 | ||
| R(C2-H7) | 1.070 | 1.081 | 1.081 | 1.081 | 1.070 | 1.081 | 1.081 | 1.081 | 1.076 | 1.091 | ||
| R(C3-H8) | 1.071 | 1.082 | 1.082 | 1.082 | 1.071 | 1.082 | 1.082 | 1.082 | 1.077 | 1.088 | ||
| R(N11-N12) | 1.078 | 1.130 | 1.105 | 1.104 | 1.078 | 1.131 | 1.106 | 1.104 | … | … | ||
| R(N11..H6) | 2.659 | 2.379 | 2.421 | 2.518 | … | … | … | … | … | … | ||
| R(N11..H8) | … | … | … | … | 3.390 | 2.857 | 3.078 | 3.750 | … | … | ||
| R(N12..H9) | … | … | … | … | 3.398 | 2.847 | 3.124 | 3.801 | … | … | ||
| θ(N12-N11-H6) | 179.9 | 180.0 | 179.1 | 179.9 | … | … | … | … | … | … | ||
| θ(N11-H6-N1) | 179.9 | 180.0 | 179.0 | 179.9 | … | … | … | … | … | … | ||
| θ(N11-H8-C3) | … | … | … | … | 129.6 | 127.5 | 129.9 | 132.5 | … | … | ||
| θ(N12-H9-C4) | … | … | … | … | 128.7 | 126.2 | 126.3 | 129.0 | … | … | ||
| θ(N1-C2-C3) | 108.2 | 107.5 | 107.8 | 107.7 | 108.2 | 107.4 | 107.7 | 107.7 | 107.7 | 107.7 | ||
| θ(C2-C3-C4) | 107.1 | 107.5 | 107.4 | 107.3 | 107.1 | 107.5 | 107.4 | 107.4 | 107.4 | 107.4 | ||
| θ(C2-N1-C5) | 109.4 | 110.1 | 109.7 | 109.9 | 109.5 | 110.1 | 109.8 | 109.9 | 109.8 | 109.8 | ||
| θ(C2-N1-H6) | 125.3 | 125.0 | 125.2 | 125.1 | 125.3 | 124.9 | 125.1 | 125.0 | 125.1 | 125.1 | ||
| θ(N1-C2-H7) | 121.1 | 121.2 | 121.0 | 121.1 | 121.1 | 121.2 | 121.1 | 121.1 | 121.5 | 126.2 | ||
| θ(C2-C3-H8) | 126.0 | 125.6 | 125.8 | 125.8 | 126.0 | 125.5 | 125.8 | 125.7 | 125.5 | 126.3 | ||
| -E (310+) | 7.75308 | 8.73940 | 9.69222 | 9.56736 | 7.75208 | 8.73785 | 9.69047 | 9.5660 | ... | … | ||
| η | 6.1749 | 5.6730 | … | … | 6.4169 | 5.9499 | … | … | … | … | ||
| μ | -1.7204 | -2.0590 | … | … | -1.5540 | -1.8722 | … | … | … | … | ||
| μM | 2.1762 | 2.3045 | 2.3384 | 2.314 | 1.8956 | 1.8954 | 1.8839 | 1.9436 | … | … | ||
| RA | 9.2888 | 9.1632 | 9.1457 | 9.1864 | 8.0718 | 7.8749 | 7.9112 | 7.9445 | … | … | ||
| RB | 0.8167 | 0.8801 | 0.8722 | 0.8451 | 0.8816 | 1.0711 | 0.9727 | 0.7643 | … | … | ||
| RC | 0.7507 | 0.8030 | 0.7963 | 0.7739 | 0.7948 | 0.9429 | 0.8662 | 0.6972 | … | … | ||
| Isomer | HF | MP2 | B3LYP | B3PW91 | ||||
| ERel | EInt | ERel | EInt | ERel | EIn | ERel | EInt | |
| Pyrrole…N2 | ||||||||
| Linear | 0.0 | -0.803 | 0.0 | -2.058 | 0.0 | -1.393 | 0.0 | -0.903 |
| T-Shaped | 0.628 | -0.176 | 0.973 | -1.086 | 1.098 | -0.282 | 0.853 | -0.044 |
| Pyrrole…CO | ||||||||
| Hydrogen bonded C end first | 0.0 | -1.512 | 0.0 | -3.150 | 0.0 | -2.711 | 0.0 | -2.146 |
| Above ring CO | +0.659 | -0.853 | +0.722 | -2.429 | +1.632 | -1.079 | +1.638 | -0.508 |
| Above ring OC | +0.559 | -0.960 | +0.753 | -2.391 | +1.650 | -1.048 | +1.682 | -0.452 |
| Hydrogen bonded O end first | +0.257 | -1.255 | +1.286 | -1.857 | +1.155 | -1.544 | +1.173 | -0.960 |
| T-shaped | +1.192 | -0.314 | +1.996 | -1.142 | +2.165 | -0.527 | +1.989 | -0.144 |
Interaction energies
Chemical hardness and chemical potential
Geometries and energies
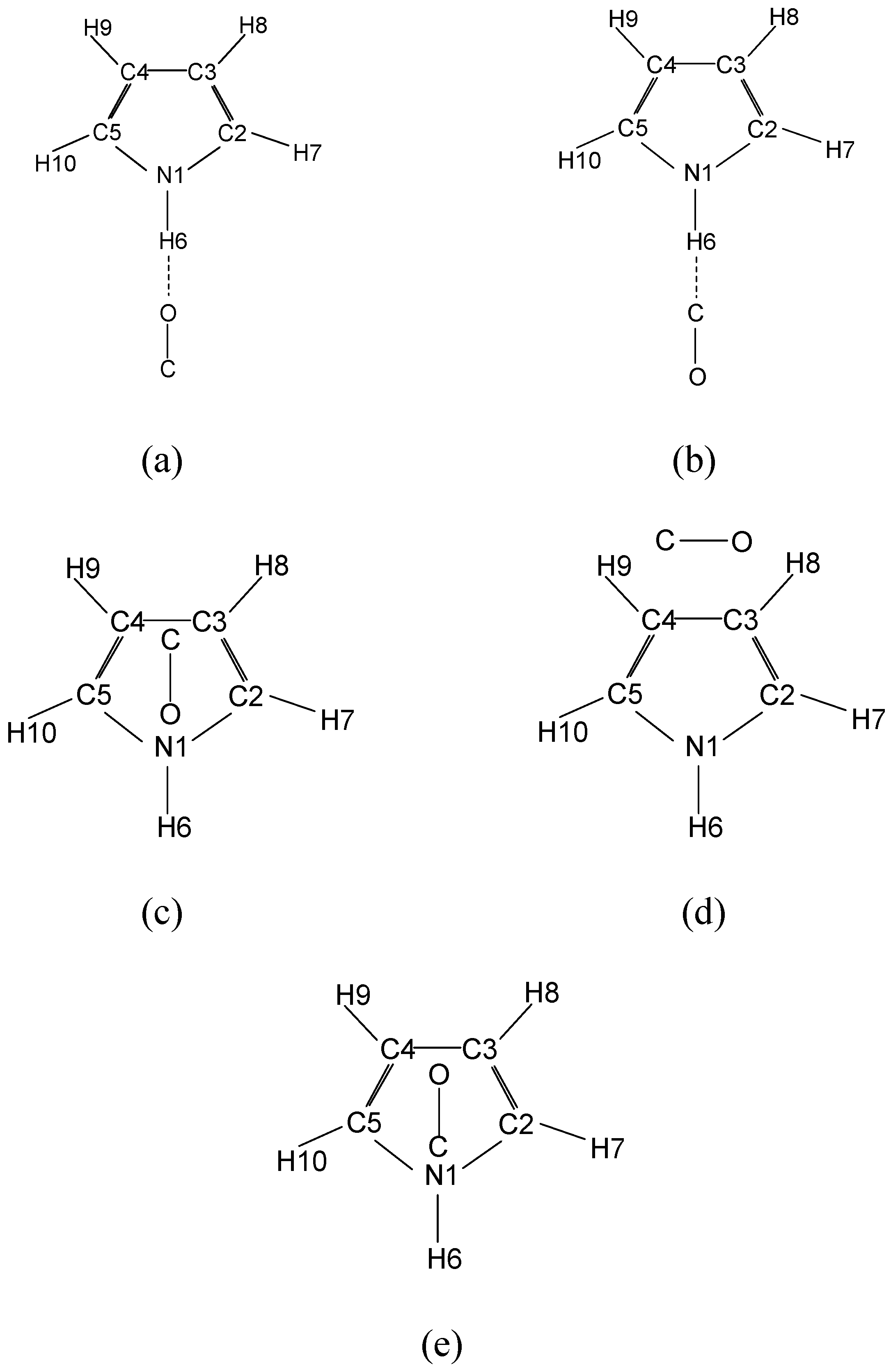
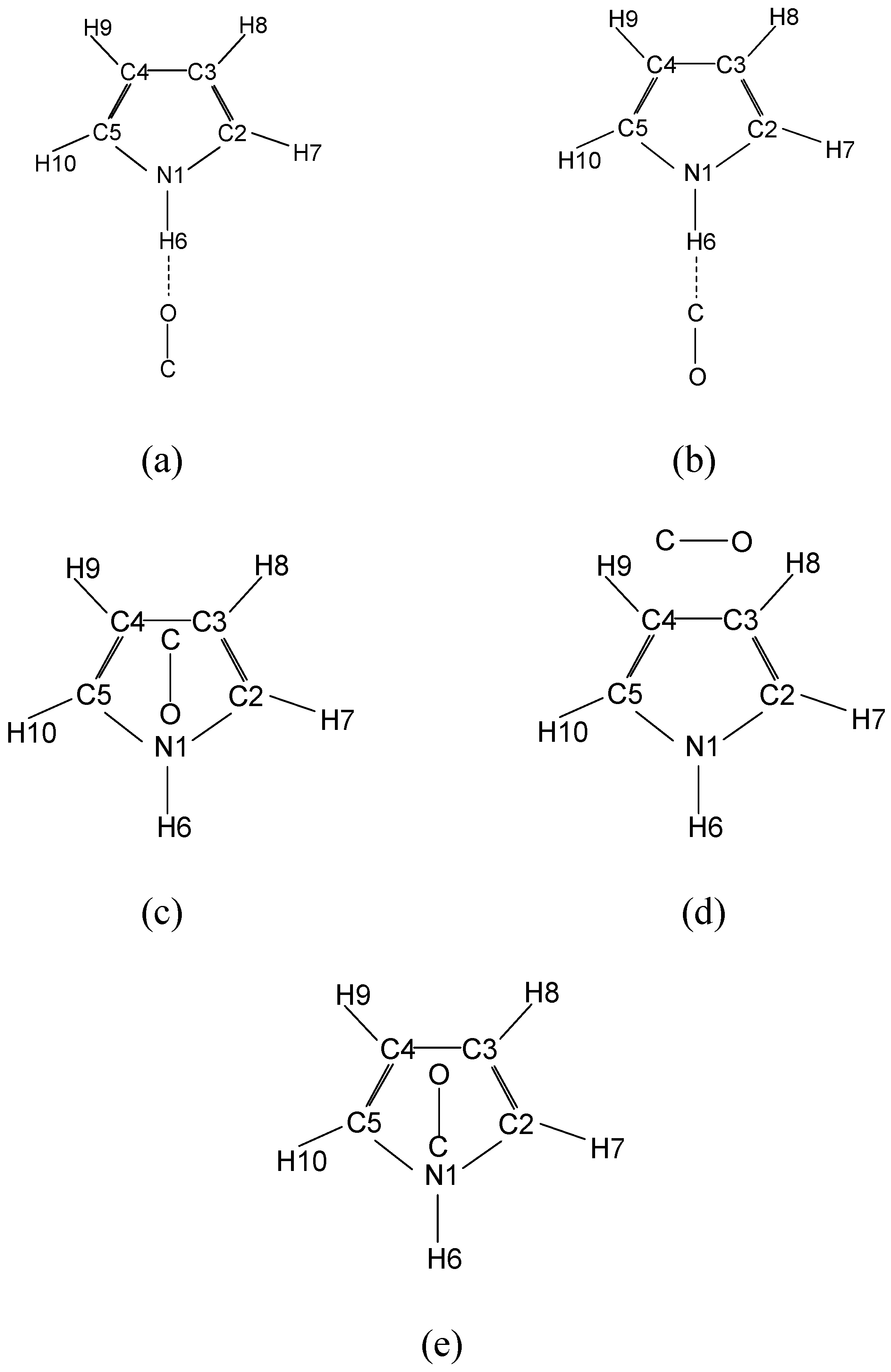
| Parameters | Hydrogen bonded O end first | Hydrogen bonded C end first | T-shaped | |||||||||||
| HF | MP2 | B3LYP | B3PW91 | HF | MP2 | B3LYP | B3PW91 | HF | MP2 | B3LYP | B3PW91 | |||
| R(N1-C2) | 1.362 | 1.373 | 1.375 | 1.370 | 1.362 | 1.372 | 1.375 | 1.370 | 1.363 | 1.373 | 1.375 | 1.371 | ||
| R(C2-C3) | 1.538 | 1.383 | 1.379 | 1.377 | 1.358 | 1.384 | 1.379 | 1.378 | 1.358 | 1.383 | 1.378 | 1.377 | ||
| R(C3-C4) | 1.426 | 1.418 | 1.425 | 1.422 | 1.426 | 1.418 | 1.425 | 1.422 | 1.427 | 1.419 | 1.426 | 1.422 | ||
| R(N1-H6) | 0.993 | 1.011 | 1.008 | 1.008 | 0.993 | 1.013 | 1.010 | 1.010 | 0.992 | 1.011 | 1.008 | 1.007 | ||
| R(C2-H7) | 1.070 | 1.081 | 1.081 | 1.081 | 1.070 | 1.081 | 1.081 | 1.081 | 1.070 | 1.081 | 1.080 | 1.081 | ||
| R(C3-H8) | 1.071 | 1.082 | 1.082 | 1.082 | 1.071 | 1.082 | 1.082 | 1.082 | 1.071 | 1.082 | 1.082 | 1.082 | ||
| R(C-O) | 1.115 | 1.152 | 1.139 | 1.138 | 1.112 | 1.149 | 1.135 | 1.135 | 1.114 | 1.151 | 1.138 | 1.137 | ||
| R(O…H6) | 2.443 | 2.295 | 2.290 | 2.404 | … | … | … | … | … | … | … | … | ||
| R(C…H6) | … | … | … | … | 2.618 | 2.400 | 2.388 | 2.416 | … | … | … | … | ||
| R(O…H8) | … | … | … | … | … | … | … | … | 3.222 | 2.864 | 3.029 | 3.129 | ||
| R(C…H9) | … | … | … | … | … | … | … | … | 3.397 | 2.925 | 3.068 | 3.485 | ||
| θ(C-O-H6) | 180.0 | 179.8 | 179.0 | 179.4 | … | … | … | … | … | … | … | … | ||
| θ(O-H6-N1) | 180.0 | 180.0 | 178.5 | 178.1 | … | … | … | … | … | … | … | … | ||
| θ(O-C-H6) | … | … | … | … | 179.7 | 180.0 | 179.5 | 179.6 | … | … | … | … | ||
| θ(C-H6-N1) | … | … | … | … | 179.7 | 180.0 | 178.8 | 178.5 | … | … | … | … | ||
| θ(O-H8-C3) | … | … | … | … | … | … | … | … | 137.5 | 131.3 | 134.5 | 137.5 | ||
| θ(C-H9-C4) | … | … | … | … | … | … | … | … | 120.6 | 123.3 | 121.4 | 121.4 | ||
| θ(N1-C2-C3) | 108.2 | 107.5 | 107.8 | 107.7 | 108.3 | 107.6 | 107.8 | 107.8 | 108.2 | 107.4 | 107.7 | 107.7 | ||
| θ(C2-C3-C4) | 107.1 | 107.5 | 107.4 | 107.3 | 107.1 | 107.4 | 107.3 | 107.3 | 107.1 | 107.5 | 107.4 | 107.4 | ||
| θ(C2-N1-C5) | 109.4 | 110.1 | 109.7 | 109.9 | 109.4 | 110.0 | 109.6 | 109.8 | 109.5 | 110.1 | 109.8 | 109.9 | ||
| θ(C2-N1-H6) | 125.3 | 125.0 | 125.1 | 125.0 | 125.3 | 125.0 | 125.1 | 125.0 | 125.3 | 124.9 | 125.1 | 125.0 | ||
| θ(N1-C2-H7) | 121.1 | 121.2 | 121.0 | 125.8 | 121.1 | 121.2 | 121.0 | 121.0 | 121.0 | 121.2 | 121.1 | 121.1 | ||
| θ(C2-C3-H8) | 126.0 | 125.6 | 125.8 | 3. | 126.0 | 125.6 | 125.6 | 125.8 | 125.9 | 125.5 | 125.7 | 125.7 | ||
| - E ( 320+) | 1.54772 | 2.50501 | 3.47779 | 3.35062 | 1.54813 | 2.50706 | 3.47963 | 3.35249 | 1.54623 | 2.50388 | 3.47618 | 3.34932 | ||
| η | 5.9779 | 5.7379 | … | … | 5.9495 | 5.6152 | 6.2379 | 5.9472 | … | … | ||||
| μ | -1.8708 | -2.0227 | … | … | -1.9306 | 2.1174 | … | … | -1.7439 | -1.8880 | … | … | ||
| μM | 2.5196 | 2.8116 | 2.3074 | 2.2559 | 2.0158 | 1.9413 | 2.5809 | 2.5850 | 1.9386 | 1.9752 | 1.8729 | 1.9354 | ||
| RA | 9.2905 | 9.1606 | 9.1456 | 9.1859 | 9.2917 | 9.1703 | 9.1540 | 9.1940 | 8.0561 | 7.8694 | 7.9209 | 7.9019 | ||
| RB | 0.8987 | 0.9301 | 0.9344 | 0.8990 | 0.8013 | 0.8435 | 0.8523 | 0.8458 | 0.9079 | 1.0494 | 0.9876 | 0.9038 | ||
| RC | 0.8194 | 0.8444 | 0.8478 | 0.8188 | 0.7377 | 0.7724 | 0.7797 | 0.7745 | 0.8159 | 0.9259 | 0.8781 | 0.8111 | ||
| Parameters | Above ring OC | Above ring CO | Experimental value | |||||||||||
| HF | MP2 | B3LYP | B3PW91 | HF | MP2 | B3LYP | B3PW91 | HF | MP2 | B3LYP | B3PW91 | |||
| R(N1-C2) | 1.362 | 1.737 | 1.375 | 1.370 | 1.362 | 1.373 | 1.374 | 1.370 | 1.370 | 1.370 | R(N1-C2) | 1.362 | ||
| R(C2-C3) | 1.358 | 1.383 | 1.378 | 1.377 | 1.358 | 1.383 | 1.379 | 1.378 | 1.382 | 1.382 | R(C2-C3) | 1.358 | ||
| R(C3-C4) | 1.427 | 1.419 | 1.426 | 1.423 | 1.427 | 1.418 | 1.425 | 1.422 | 1.417 | 1.417 | R(C3-C4) | 1.427 | ||
| R(N1-H6) | 0.993 | 1.011 | 1.008 | 1.007 | 0.993 | 1.011 | 1.008 | 1.007 | 0.996 | 1.000 | R(N1-H6) | 0.993 | ||
| R(C2-H7) | 1.070 | 1.081 | 1.080 | 1.081 | 1.070 | 1.081 | 1.080 | 1.081 | 1.076 | 1.091 | R(C2-H7) | 1.070 | ||
| R(C-O) | 1.115 | 1.152 | 1.139 | 1.138 | 1.114 | 1.151 | 1.139 | 1.138 | … | … | R(C-O) | 1.115 | ||
| R(O…N1) | 3.569 | 3.171 | 3.402 | 3.854 | … | … | … | … | … | … | R(O…N1) | 3.569 | ||
| R(C…N1) | … | … | … | … | 3.780 | 3.157 | 3.441 | 3.533 | … | … | R(C…N1) | … | ||
| R(O…C2) | 3.786 | 3.375 | 3.582 | 3.940 | … | … | … | … | … | … | R(O…C2) | 3.786 | ||
| R(C…C2) | … | … | … | … | 3.778 | 3.357 | 3.463 | 3.556 | … | … | R(C…C2) | … | ||
| R(O…C5) | 3.786 | 3.375 | 3.583 | 3.887 | … | … | … | … | … | … | R(O…C5) | 3.786 | ||
| R(C…C5) | … | … | … | … | 3.770 | 3.357 | 3.460 | 3.561 | … | … | R(C…C5) | … | ||
| θ(O-N1-C2) | 88.5 | 86.4 | 86.1 | 71.8 | … | … | … | … | … | … | θ(O-N1-C2) | 88.5 | ||
| θ(O-N1-C5) | 88.4 | 86.4 | 86.2 | 81.2 | … | … | … | … | … | … | θ(O-N1-C5) | 88.4 | ||
| θ(C-N1-C2) | … | … | … | … | 79.5 | 86.1 | 79.4 | 79.8 | … | … | θ(C-N1-C2) | … | ||
| θ(C-N1-C5) | … | … | … | … | 79.2 | 86.1 | 79.3 | 80.0 | … | ... | θ(C-N1-C5) | … | ||
| θ(N1-C2-C3) | 108.2 | 107.4 | 107.7 | 107.7 | 108.2 | 107.4 | 107.7 | 107.7 | 107.7 | 107.7 | θ(N1-C2-C3) | 108.2 | ||
| θ(C2-C3-C4) | 107.1 | 107.5 | 107.4 | 107.4 | 107.1 | 107.5 | 107.4 | 107.3 | 107.4 | 107.4 | θ(C2-C3-C4) | 107.1 | ||
| θ(C2-N1-C5) | 109.5 | 110.2 | 109.8 | 110.0 | 109.5 | 110.2 | 109.9 | 110.0 | 109.8 | 109.8 | θ(C2-N1-C5) | 109.5 | ||
| θ(C2-N1-H6) | 125.2 | 124.9 | 125.1 | 125.0 | 125.2 | 124.9 | 125.0 | 125.0 | 125.1 | 125.1 | θ(C2-N1-H6) | 125.2 | ||
| θ(N1-C2-H7) | 121.1 | 121.2 | 121.1 | 121.1 | 121.1 | 121.2 | 121.1 | 121.1 | 121.5 | 126.2 | θ(N1-C2-H7) | 121.1 | ||
| θ(C2-C3-H8) | 126.0 | 125.6 | 125.8 | 125.8 | 126.0 | 125.6 | 125.8 | 125.8 | 125.5 | 126.3 | θ(C2-C3-H8) | 126.0 | ||
| -E(320+) | 1.54724 | 2.50586 | 3.47700 | 3.34981 | 1.54708 | 2.50591 | 3.47703 | 3.34988 | … | … | -E(320+) | 1.54724 | ||
| η | 6.4693 | 6.2548 | … | … | 6.0285 | 6.2258 | … | … | … | … | η | 6.4693 | ||
| μ | -1.5655 | -1.6571 | … | … | -2.1776 | -1.7111 | … | … | … | … | μ | -1.5655 | ||
| μM | 1.6065 | 1.5201 | 1.9379 | 2.0099 | 2.1918 | 2.2042 | 1.9358 | 1.9385 | … | … | μM | 1.6065 | ||
| RA | 4.3337 | 4.2135 | 4.2129 | 4.2345 | 4.4545 | 4.2074 | 4.3764 | 4.3751 | … | … | RA | 4.3337 | ||
| RB | 1.5978 | 1.9415 | 1.7790 | 1.5561 | 1.4423 | 1.8846 | 1.6087 | 1.5519 | … | … | RB | 1.5978 | ||
| RC | 1.5599 | 1.8733 | 1.7227 | 1.5128 | 1.4239 | 1.8190 | 1.5840 | 1.5265 | … | … | RC | 1.5599 | ||
Interaction energies
Chemical hardness and chemical potential
Conclusion
Acknowledgments
References
- Hobza, P.; Zahradnik, R. Chem. Rev. 1988, 88, 871.
- Chapman, D. M.; Müller-Dethlef, K. J. Chem. Phys. 1999, 111, 1955, and references there in.
- Webber, T.; Smith, A. M.; Riedle, E.; Neusser, H. J.; Schlag, E.W. Chem. Phys. Lett. 1990, 175, 79. [CrossRef]
- Brupbacher, T.; Bauder, A. J. Chem. Phys. 1993, 99, 9394.
- Hu, Y. H.; Lu, W. Y.; Yang, S. H. J. Chem. Phys. 1996, 105, 5305. [CrossRef]
- Brookes, M. D.; McKeller, A. R. W. J. Chem. Phys. 1998, 109, 5823. [CrossRef]
- Altman, R. S.; Marshal, M. D.; Klemperer, W. J. Chem. Phys. 1983, 79, 57. [CrossRef]
- Broquier, M.; Chevalier, M.; Picard-Bersellini, A. Mol. Phys. 1997, 91, 123.
- Goodwin, E. J.; Legon, A. C. J. Chem. Phys. 1985, 82, 4434.
- Lundell, J.; Räsänen, M. J. J. Chem. Phys. 1998, 109, 5823.
- Hains, S. R.; Dessent, C. E. H.; Müller-Dethlef, K. J. Chem. Phys. 1999, 111, 1947.
- Coblentz, W. W. Investigation of Infra-red Spectra; Carnegie Institute: Washington, DC, 1905; p. 331 p. [Google Scholar]
- Maroni-Ansidei, R.; Rolla, M. Atti. Accad. Lincei. 1938, 27, 410.
- Lord, R. C.; Miller, F. A. J. Chem. Phys. 1942, 10, 328.
- Mirone, P. Gazz. Chim. Ital. 1956, 86, 165.
- Sicorov, N. K.; Kalashnikova, L. P. Opt. Spectrosc. 1968, 26, 247.
- Lautié, A.; Novak, A. J. Chim. Phys. 1972, 69, 1332.
- Scott, D. W. J. Mol. Spectrosc. 1971, 37, 77. [CrossRef]
- Geidel, E.; Billes, F. J. Mol. Struct.(Theochem) 2000, 507, 75. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.M.W.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.; Petersson, G.A.; Montgomery, J.A.; Raghavachari, K.; Al-Laham, M.A.; Zakrzewski, V.G.; Ortiz, J.V.; Foresman, J.B.; Cioslowski, J.; Stefanov, B.B.; Nanayakkara, A.; Challacombe, M.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andres, J.L.; Replogle, E.S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J.S.; Defrees, D.J.; Baker, J.; Stewart, J.P.; Head-Gordon, M.; Gonzalez, C.; Pople, J.A. Gaussian 94, Revision E.2; Gaussian: Pittsburgh PA, 1995. [Google Scholar]
- Becke, A. D. Phys. Rev. A 1988, 38, 3098. [CrossRef]
- Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B. 1988, 37, 785. [CrossRef]
- Perdew, J. P.; Wang, Y. Phys. Rev. B 1992, 45, 244. [CrossRef]
- Ghosh, S. K.; Berkowitz, M. J. Chem. Phys. 1985, 83, 2976.
- Berkowitz, M.; Ghosh, S. K.; Parr, R. G. J. Am. Chem. Soc. 1985, 107, 6811. [CrossRef]
- Paizs, B.; Suhai, S. J. Comp. Chem. 1998, 19, 575.
- Boys, S. F.; Bernardi, F. Mol. Phys. 1970, 19, 553.
- Nygaard, L.; Nielsen, J. T.; Kirchheiner, J.; Maltsen, G.; Rastrup-Andersen, J.; Sorensen, G. O. J. Mol. Struct. 1969, 3, 491. [CrossRef]
- Xie, Y.; Fan, K.; Boggs, J. E. Mol. Phys. 1986, 58, 401.
- Curtiss, L. A.; Eisgruber, C. L. J. Chem. Phys. 1984, 80, 2022.
- Wesolowski, T. A.; Parisel, O.; Ellinger, Y.; Weber, J. J. Phys. Chem. A. 1997, 101, 7818. [CrossRef]
- Pearson, R. G. J. Chem. Ed. 1987, 64, 561. [CrossRef]
- Parr, R. G.; Chattaraj, P. K. J. Am. Chem. Soc. 1991, 113, 1854. [CrossRef]
- Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512. [CrossRef]
- Bartell, L. S. J. Chem. Ed. 1968, 45, 754. [CrossRef]
- Burdett, J. K.; Coddens, B. K.; Kulkarni, G. V. Inorg. Chem. 1988, 27, 3259.
- Pearson, R. G. Chemtracts Inorg. Chem. 1991, 3, 317.
- Chattaraj, P. K. J. Indian Chem. Soc. 1992, 69, 173.
- Parr, R. G.; Chattaraj, P. K. J. Am. Chem. Soc. 1991, 96, 3283.
- Chattaraj, P. K.; Schilyer, P. V. R. J. Am. Chem. Soc. 1993, 116, 1069.
- Chattaraj, P. K.; Lee, H.; Parr, R. G. J. Am. Chem. Soc. 1991, 113, 1855. [CrossRef]
- Pearson, R. G. Proc. Natl. Acad. Sci. USA. 1988, 82, 6723.
- Nath, S.; Sannigrahi, A. B.; Chattaraj, P. K. J. Mol. Struct. (Theochem) 1994, 309, 65. [CrossRef]
- Arulmozhiraja, S.; Kolandaivel, P. Int. J. Quant. Chem. 1997, 64, 231.
- Graham, C.; Impie, D. A.; Raab, R. E. Mol. Phys. 1998, 93, 49.
- Lundell, J.; Latajka, Z. J. Phys. Chem. 1997, 101, 5004. [CrossRef]
- Legon, A. C.; Millen, D. J. Acc. Chem. Res. 1987, 20, 39. [CrossRef]
- Hobza, P.; Šponer, J.; Reschel, T. J. Comput. Chem. 1995, 16, 1315. [CrossRef]
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
Kanakaraju, R.; Kolandaivel, P. Post Hartree–Fock and DFT Studies on Pyrrole···Nitrogen and Pyrrole···Carbon Monoxide Molecules. Int. J. Mol. Sci. 2002, 3, 777-789. https://doi.org/10.3390/i3070777
Kanakaraju R, Kolandaivel P. Post Hartree–Fock and DFT Studies on Pyrrole···Nitrogen and Pyrrole···Carbon Monoxide Molecules. International Journal of Molecular Sciences. 2002; 3(7):777-789. https://doi.org/10.3390/i3070777
Chicago/Turabian StyleKanakaraju, R., and P. Kolandaivel. 2002. "Post Hartree–Fock and DFT Studies on Pyrrole···Nitrogen and Pyrrole···Carbon Monoxide Molecules" International Journal of Molecular Sciences 3, no. 7: 777-789. https://doi.org/10.3390/i3070777
APA StyleKanakaraju, R., & Kolandaivel, P. (2002). Post Hartree–Fock and DFT Studies on Pyrrole···Nitrogen and Pyrrole···Carbon Monoxide Molecules. International Journal of Molecular Sciences, 3(7), 777-789. https://doi.org/10.3390/i3070777