2.1 Preliminaries
We begin this section with a very brief summary of the essential ingredients of the SS-MRCC formalism. This will form the starting point for the perturbative approximations to follow. We write the reference function
as a combination of the reference determinants
spanning the CAS:
The exact function
is written as a cluster expansion involving cluster operators
exciting from corresponding
’s:
is taken to satisfy the Schrödinger equation with the eigenvalue
E:
Each
excites to all the virtual functions from
via the various
n hole –
n particle excitations, where the holes and particles are defined with respect to each
. Such a cluster expansion Ansatz was first used by Jeziorski and Monkhorst in the context of the effective hamiltonian based state-universal multi-reference coupled-cluster (SU-MRCC) theory [
2] and has later been exploited in the state-specific formulations too [
9,
10,
36]. Since each
has different sets of active orbitals, any specific core-to-particle excitation would lead to a different virtual determinant from each
. This is, however, not so in general for excitations involving active orbitals. Thus, we would encounter
redundancy of the cluster operators involving active orbitals. To determine all of them, we have to invoke suitable sufficiency conditions. One may imagine that sufficiency conditions introduce a great degree of arbitrariness in a formalism. This is, however, not so if we want to exploit the arbitrariness in our choice to satisfy our twin desirable goals: to ensure that intruders are absent and to guarantee size-extensivity. It has been found that there are only two choices which naturally lead to MRCC equations which generate manifestly connected cluster operators. One set is just the SU-MRCC theory of Jeziorski and Monkhorst [
2], which is known to encounter intruders. The other is our SS-MRCC formalism [
9,
10]. We present below, without the detailed derivation, the form of the working equations for the cluster amplitudes:
where
and
The model space coefficients
are determined from
The sets
and
are coupled through eq. (4) and eq. (7). Solving these coupled set of equation gives us the cluster amplitudes and the converged coefficients from the diagonalization.
For the detailed derivation and the proof of the extensivity of the SS-MRCC theory we refer to our recent papers [
9,
10]. What is pertinent for us here is the identification of one of the essential arguments leading to extensivity, since this will form the guideline of the perturbative approximations to follow. Dividing eq. (4) through by
, we have
The first term of eq. (8) above is manifestly extensive, while the connectivity property of the second term requires a careful treatment, since this involves a product of two matrix-elements and may not have terms with common orbital labels in the two factors. Using the Baker-Campbell-Hausdorff formula for the product of exponentials, the second term can be written as
Now, the second factor in eq. (9),
is labeled by all the active orbitals which distinguish the determinants
and
, and the first factor
should contain terms with some of these distinguishing active orbitals in
for extensivity. While it is straightforward to show that the commutators and the multiple commutators generated by the Baker-Campbell-Hausdorff formula do have active orbital labels with this property, the individual terms linear in
and
do not. In fact there are excitation operators involving orbitals different from those active orbitals distinguishing
and
. It was proved by us [
9,
10] that the term
containing
the difference is, however, labeled by some or all the active orbitals distinguishing
and
, and thus the two factors in the term in eq. (9) above have indeed some orbital labels in common. For any approximation of the SS-MRCC equations preserving the extensivity,
it is mandatory to treat all the cluster amplitudes on an equal footing; otherwise the difference
will not be labeled by the active orbitals distinguishing
and
. This aspect forms one guiding principle in our development of the perturbative approximations.
2.2 State-specific multi-reference perturbation theories: SS-MRPT
We wish to view the low order perturbative versions as a suite of quasi-linearized approximations of the SS-MRCC theory. Towards this end, we rewrite the leading terms of the cluster amplitude finding equations, eq. (4), of the parent SS-MRCC theory in the following form:
The four distinct terms in the above expression are separately shown under four brackets. The first term essentially corresponds to the coupling of a virtual function to a model function, and is akin to the numerator in a simple perturbation theory. The second term is a commutator of
and
, and with
approximating
contributes an RS-like denominator of a traditional effective hamiltonian-based theory. The third and the fourth terms together perform two inter-related but distinct functions: (a) to convert the usual RS-like denominators into one containing the actual state energies, to bypass intruders – as befitting a state-specific theory, and (b) to supply counter-terms guaranteeing size-extensivity of the theory. The third term, in fact, supplies the term containing the state-energy, as shown below, while the fourth term, which couples the different model functions via the dressed hamiltonian
containing
, is, in conjunction with the third term, responsible to maintain size-extensivity.
Let us first briefly review our former perturbative formulations. This will help not only in emphasizing certain theoretical issues which any perturbative approximant has to satisfy, but also will serve to indicate where a more flexible approach can be taken. We recall at this point the observation noted earlier (after eq. (9)) that the term is connecte provided and are treated on the same footing. This aspect has a direct bearing on the structure of the RS and BW form of the working equations in any size-extensive perturbative formalism, viz. the last two terms in eq. (10) should be treated in the same approximation.
In the original formulation, we treated all the four terms consistently in the same partitioning scheme. This led to a rather inflexible approach, since this necessarily constrained us to use only a very specific partitioning strategy. Since it is natural to have the unperturbed state-energy
appear in the denominator in the RS version, we approximated
by
in
, since this leads to:
To treat the term containing
on the same footing, it should thus appear multiplied by
in the RS version. In a consistent perturbative approach, each term should be of first order in a first order RS formulation. Thus
in the last two terms in eq. (10) should be interpreted as
. The partitioning of
H in this approach is thus dictated by the necessity of keeping the full active portion of the hamiltonian in
. For the definition of
for the virtual functions, it is natural to choose it as in the traditional EN partition. We thus advocated the following strategy in our earlier formulation: we partitioned the hamiltonian,
H, into an unperturbed part,
, and a perturbation,
V. We used a multi-partition strategy in that the unperturbed
was chosen as dependent on the
it acts upon, analogous to what was advocated in [
33].
is a sum of
, the diagonal part of the Fock operator,
, with respect to
as vacuum, when there is at least one inactive orbital, the whole active block of
, plus all the ladder operators of the two-body term which contains at least one inactive orbital and the entire active portion of the two-body term. Though this resembles the choice of Dyall [
28] in the context of CASPT2, it is appropriately generalized in the context of multi-partitioning.
The eq. (10) was expanded in orders of perturbation to systematically generate the proper RS and BW versions of the perturbative expansion. While the RS version used E as a power series expansion, in the BW the E was kept unexpanded. We expanded each cluster operator that appear in the above equation as a power series in V. The same approximations were invoked while computing the third and the fourth terms. We should note here that in this formulation is non-diagonal in the active orbitals, which leads to coupling of various amplitudes of in the second term of eq. (10)
Instead of a strict perturbative analysis of all the four terms in the quasi linearized SS-MRCC theory, we want now to treat the third and the fourth terms of eq. (10) as something to be computed independently of the perturbative order. To motivate towards further development, we rewrite the third term explicitly in terms of the ‘state-energy’
:
In the above expression, we treat
as dependent on our choice of
, depending on the RS or BW mode of formulation, but not on a specific partitioning strategy. We choose
simply as
for the RS version, or as the second order effective pseudo-operator,
for the BW version. The partitioning of
H affects only the terms
and
. Since the partitioning of
H and the treatment of the size-extensivity correction term are independent now, we can choose
to be even a one-particle operator, reminiscent of a truly MP theory. We can also envision using an EN type of partition for
H. In both the choices,
is a diagonal operator, and this lends a simpler structure to our new perturbation theory. Expanding the first two terms of eq. (12) in orders of perturbation, and retaining only the terms of the first order, we have
In our MP partition, we choose
to be a sum of the Fock operator for the function
. This will correspond again to a multi-partitioning MP perturbation theory [
33]. In the EN case,
contains in addition all the diagonal direct and exchange ladders. In this paper we will explore the efficacy of the new multi-partitioned MP and EN type formulations only, as proposed above.
For actual applications, and to emphasize the organizational aspects of the theory, we rewrite the working equations, eq. (12), in the following form:
We note that the only coupling between the various
Ts are via the sum over
ν appearing in the numerator of eq. (14) above. There is thus no coupling between the various excitation components in
, and the coupling is present with only those
which lead to the same excitation as by the product of excitation operators for the specific
under consideration. This leads to a very attractive computational scheme, where we consider each type of excitation involving a specific set of orbitals, and compute all the
T amplitudes for various
µ with the same set of orbitals using eq. (14). The contributions of all these
T amplitudes to the effective pseudo-operator
are then computed, and a fresh set of excitations considered next. Thus, no
T amplitudes need to be stored in this formulation, and the coupling is minimal.
It is interesting to compare the working equations of MRMP [
26,
27] and our SS-MRPT. Because of the sufficiency conditions stemming from the redundancy in our formulation, the projection on to the various virtual functions has to be considered for each model function. The minimal coupling in eq. (14) above take care of both the redundancy and size-extensivity. The solution of these equations require the storage of only those
amplitudes for various
µs which are labeled by the same spin-orbitals. In the first step of the iteration, if the couplings are ignored, we get almost the same working equations of the MRMP theory. Since in the MRMP formulation, the denominator for each
also requires a separate calculation, the extra work entailed in our theory as compared to the MRMP theory is not significantly large. Thus, by paying some extra computational price, we could ensure the rigorous size-extensivity of our formulation.
As emphasized earlier, for the RS theories
, corresponding to the CAS energy and the term
is just
. For the BW version
, the second-order energy obtained by diagonalizing
. In both cases
is given by
The second order energy
is obtained from
For the MP partitioning, the quantity
would be the difference of the diagonal elements of
containing the occupied and unoccupied orbitals of
involved in the excitation. For the EN partition, the corresponding term will involve, in addition, the diagonal direct and exchange ladders involving the same orbitals.
The eqs. (14), (15) and (16) are our principal working equations. It is noteworthy that in the SS-MRPT(RS) formalism the zeroth order coefficients ’s are used to evaluate the cluster operators in eq. (14), but the coefficients are relaxed during the computation of , since this is obtained by diagonalization via eq. (16). On the other hand, in the BW context, the coefficients are iteratively updated.
The robustness of the energy denominators in the presence of intruders is quite manifest in our SS-MRPT formalisms: the denominators are of the form . The difference is usually smaller than the term . The latter is never small as long as the unperturbed or the perturbed energy, , is well-separated from the energies of the virtual functions. This holds true even if some of the ’s are close to . The above arguments remain equally valid even in the case of EN partitioning. In this case, for the RS version, , and , and the denominator takes the simple form . Both the perturbation theories are thus intruder-free, and both are explicitly size-extensive. They are also size-consistent when we use orbitals localized on the separated fragments.
The above development has been in terms of spin-orbitals. The spin-adaptation of SS-MRCC or SS-MRPT for states of arbitrary spins is rather non-trivial, and requires quite extensive formal developments. We shall present the spin-adapted version in our future publications. For the CAS involving only closed-shell singlets, however, the spin-adaptation is very simple: we replace the spin-orbital indices by orbital indices, and assign a factor of 2 for each ‘loop’ (when the terms are expressed diagrammatically). These types of model spaces are the ones we will use in our applications, and will work with the spin-adapted expressions.

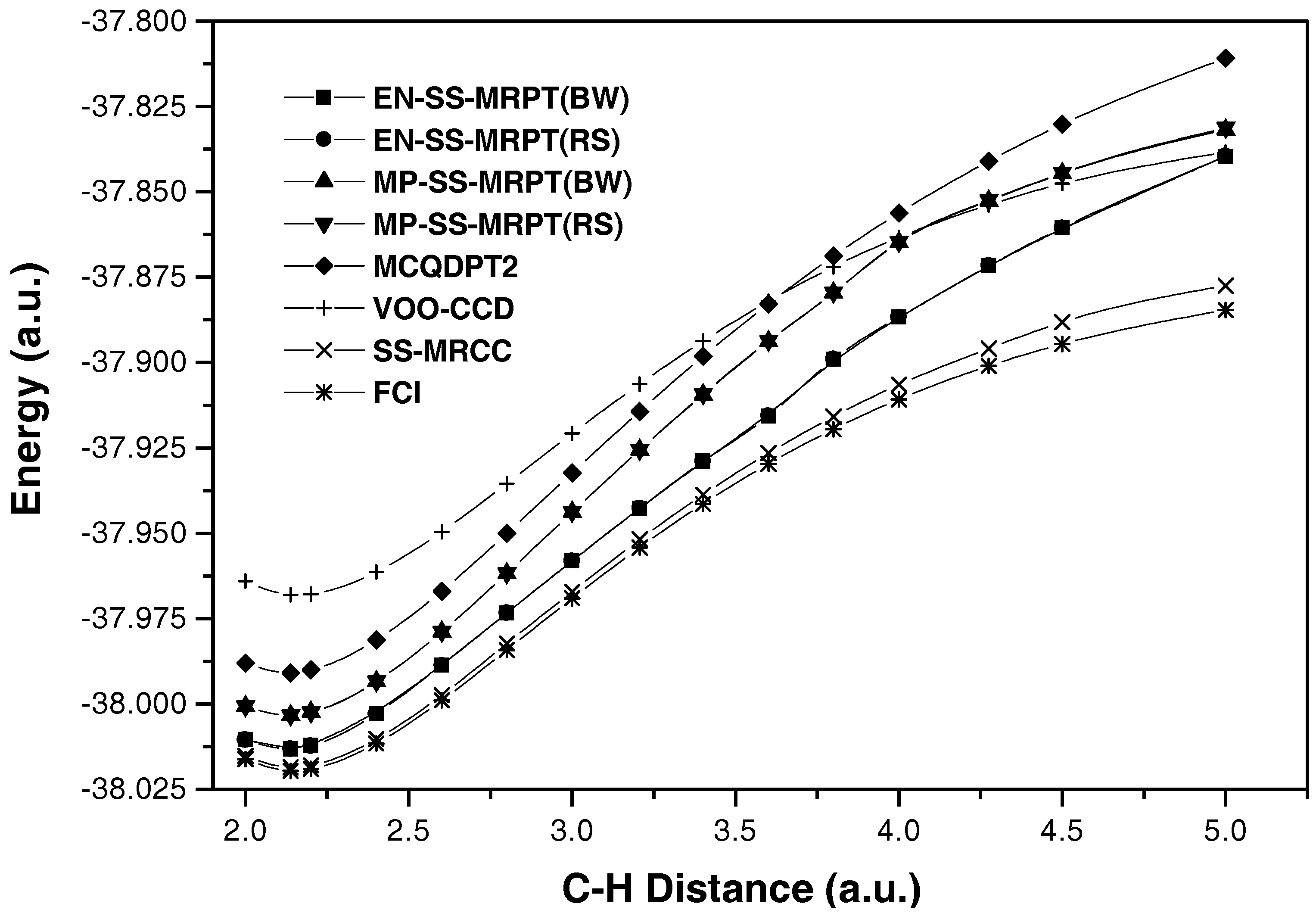

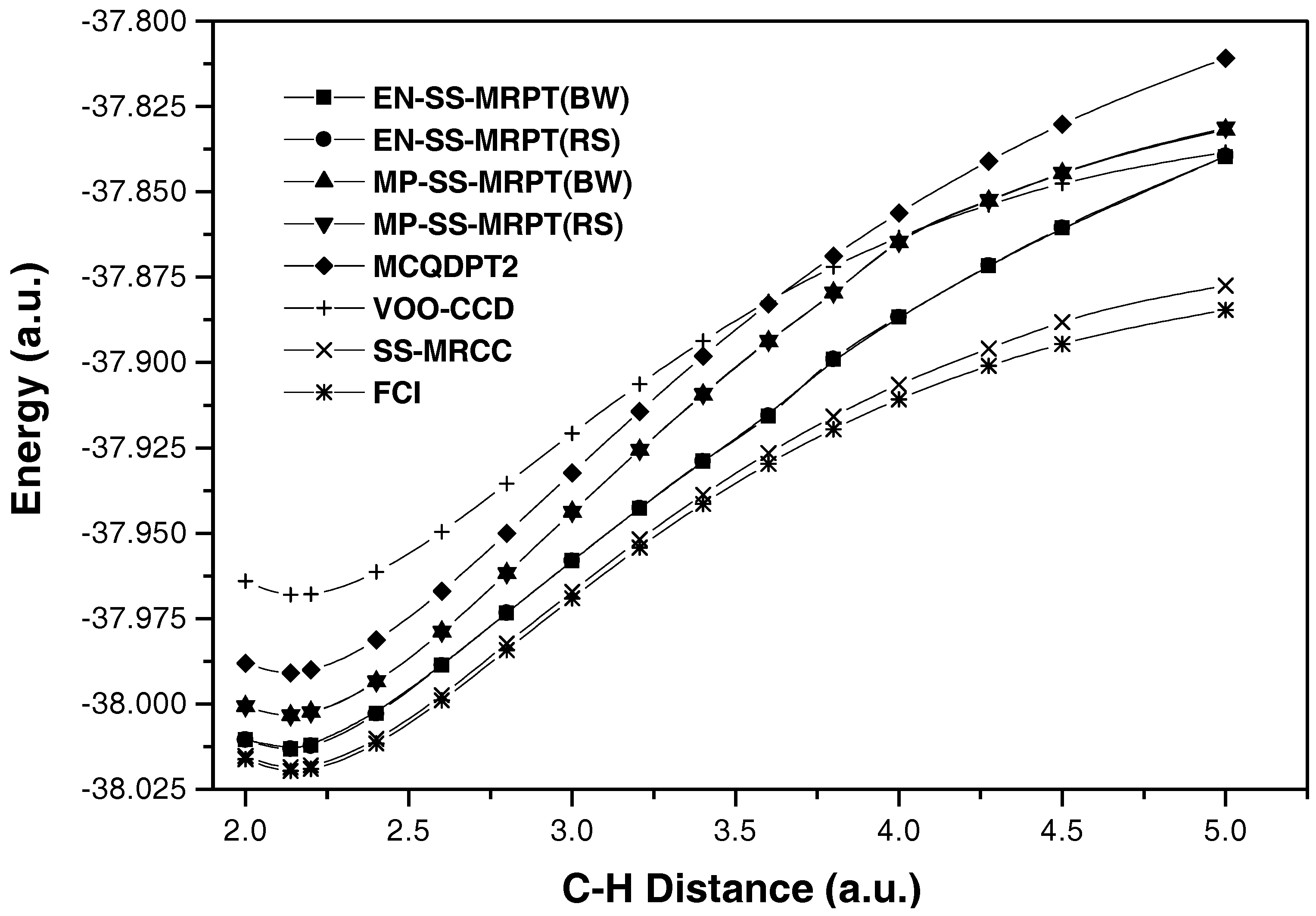{kind=link}
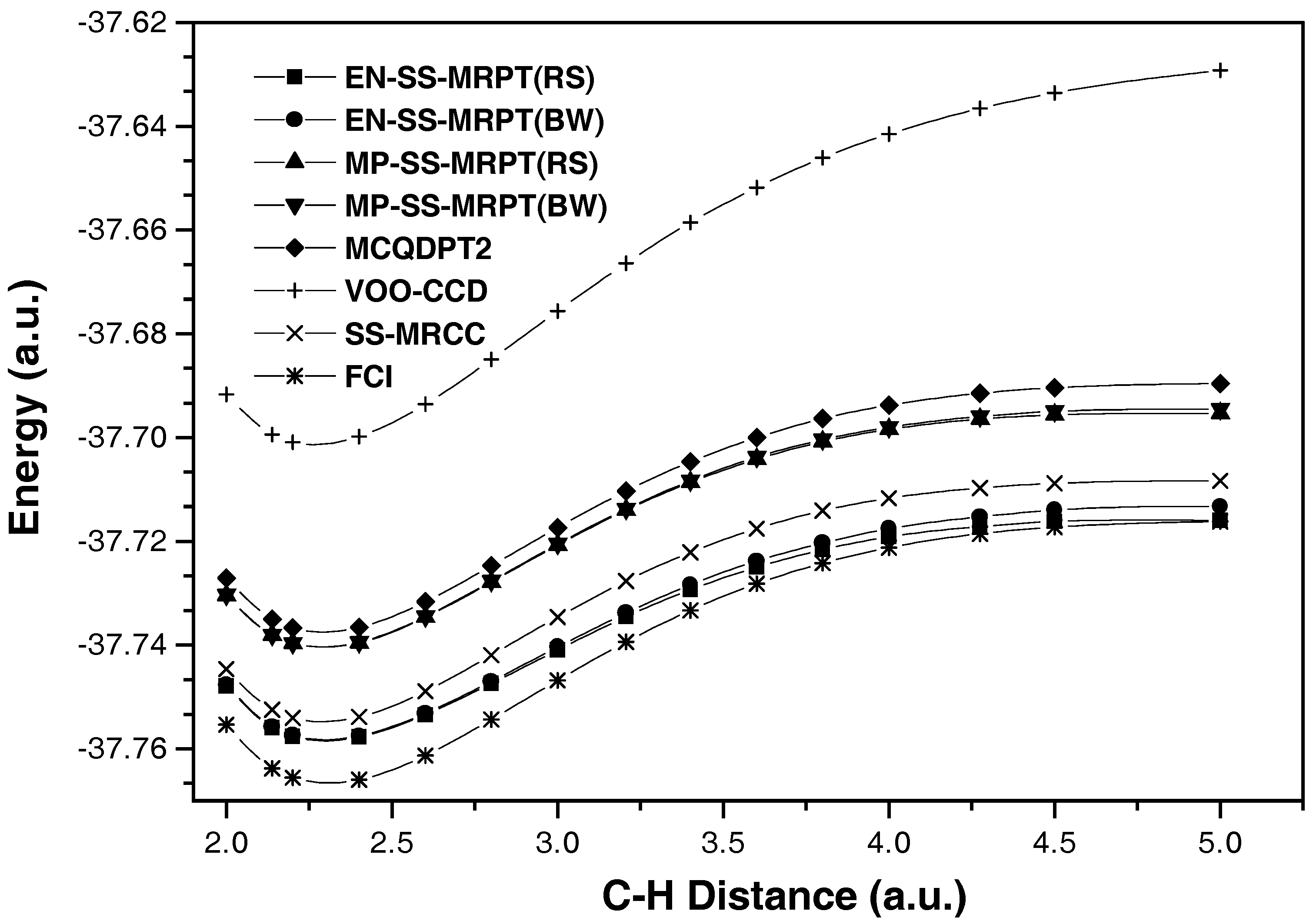
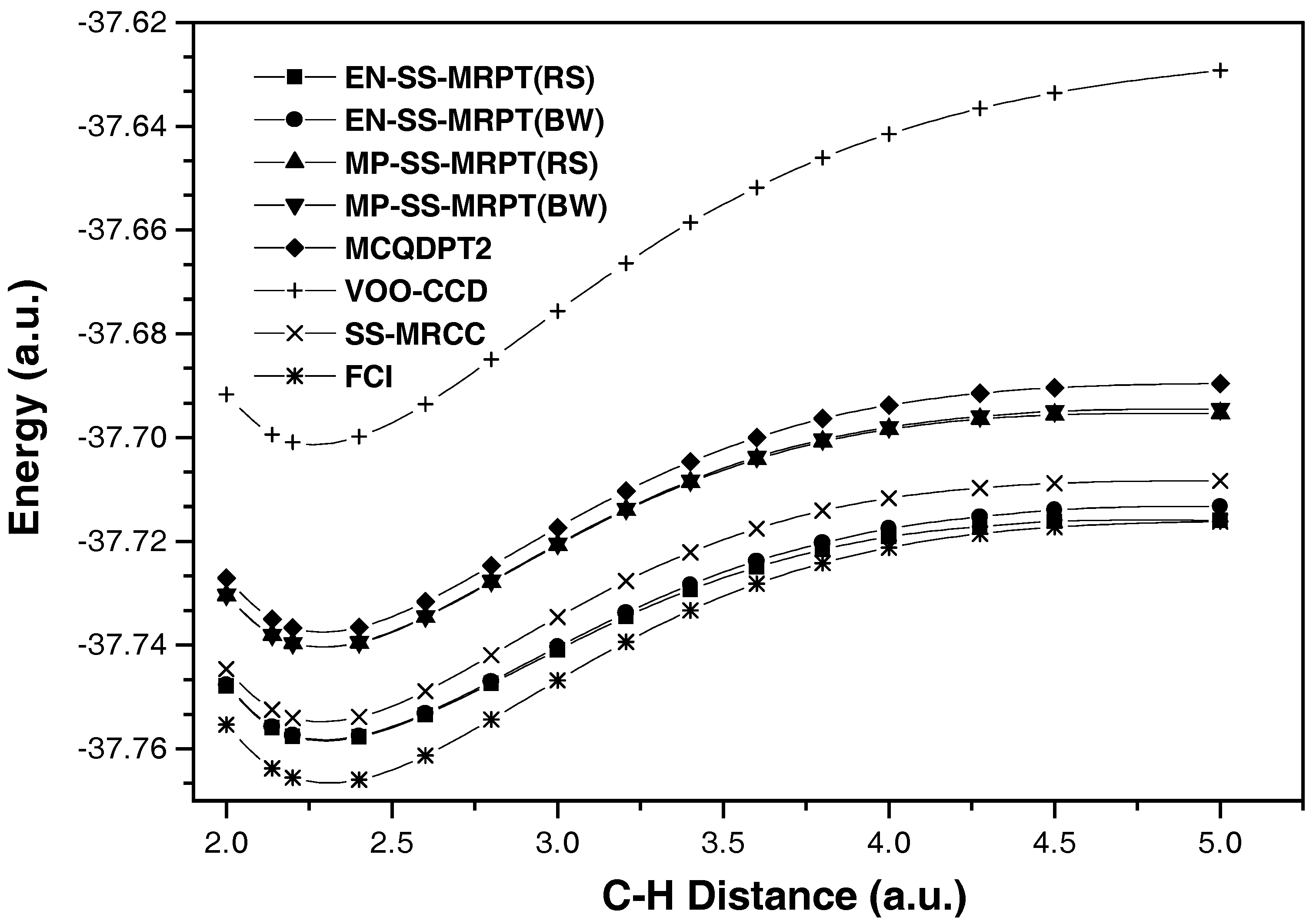{kind=link}