
Pan-Cancer Analysis Identifies a Ras-Related GTPase as a Potential Modulator of Cancer

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
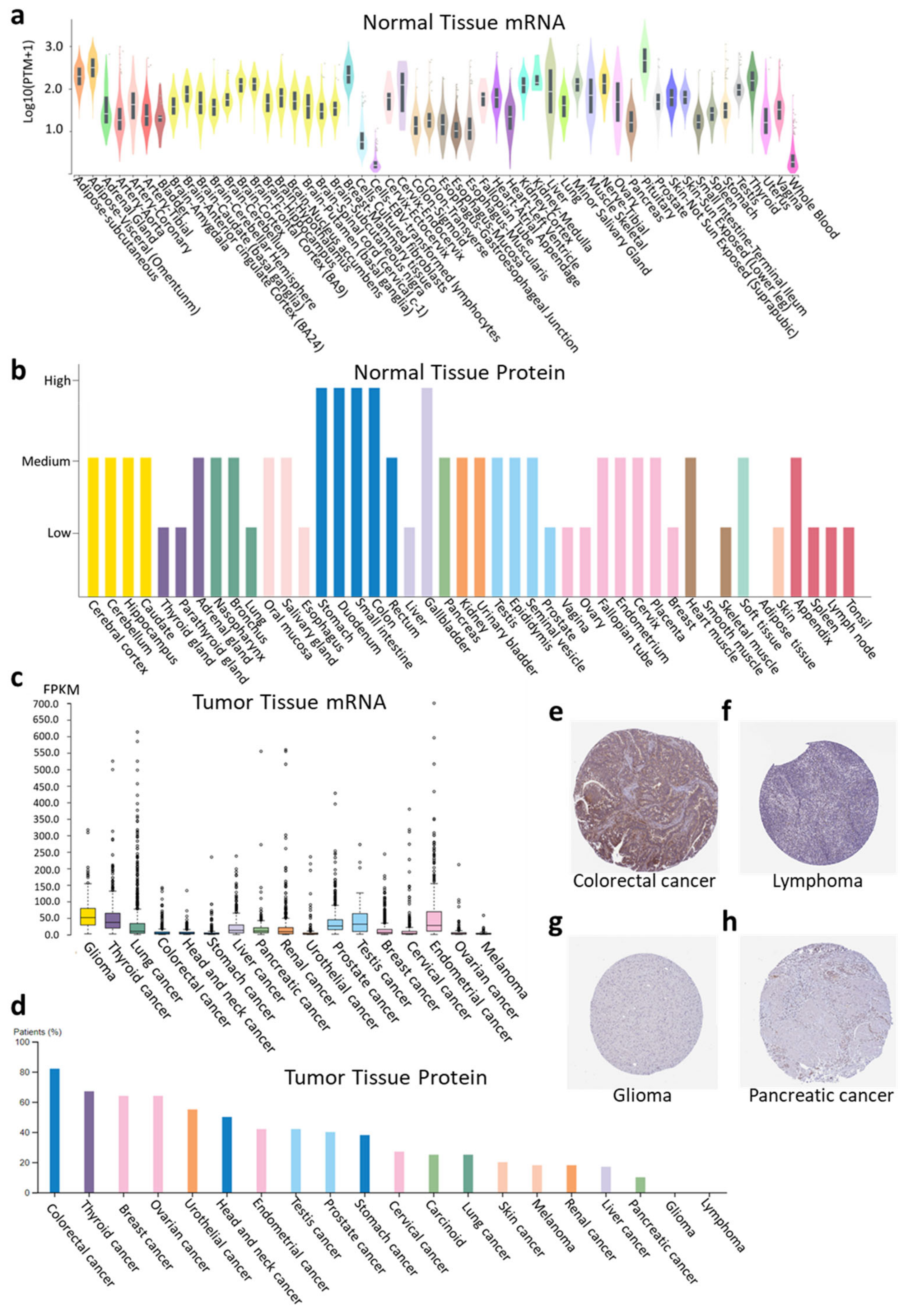
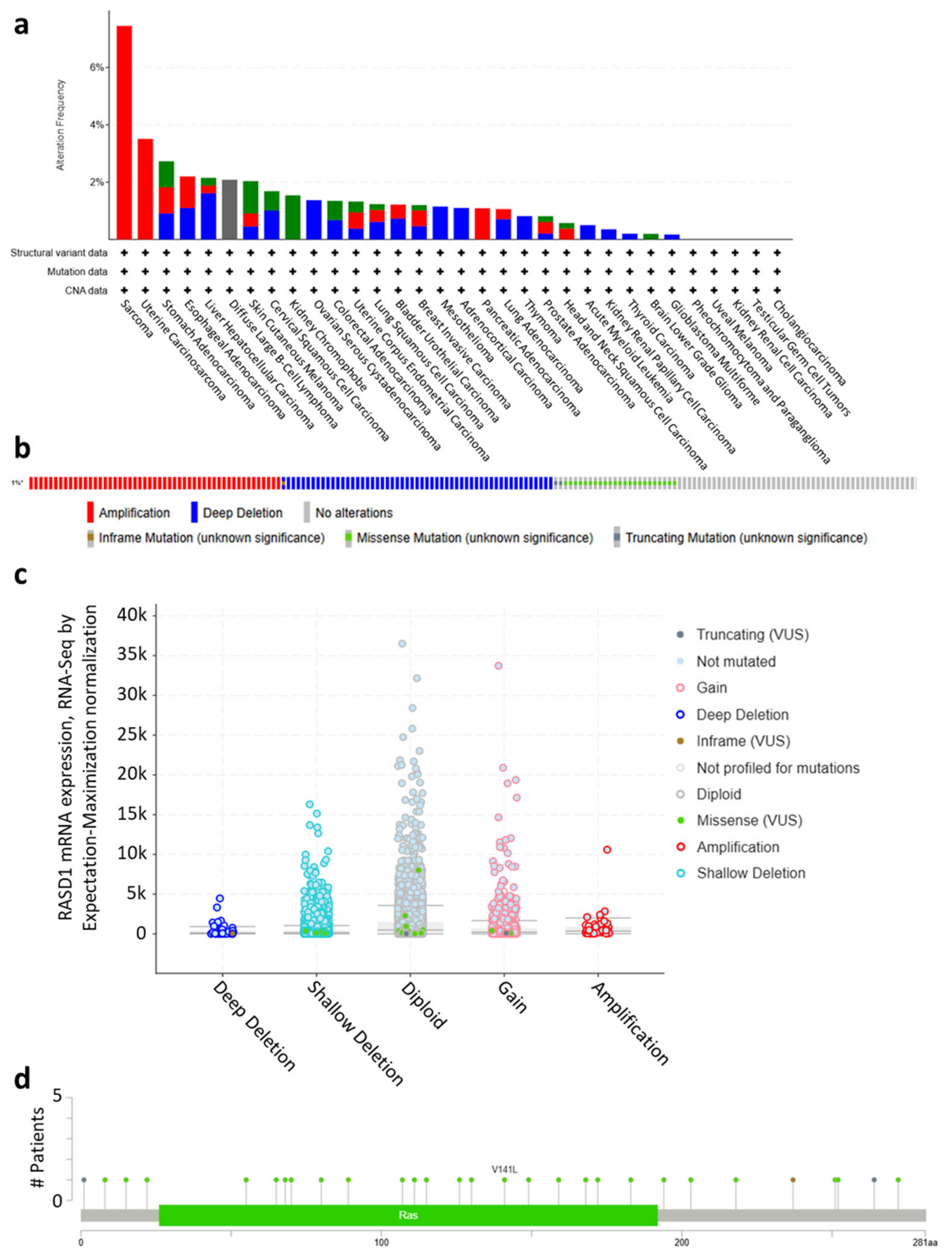
2.1. RASD1 Is Expressed in Normal and Cancer Tissues but Is Generally Downregulated in Cancer
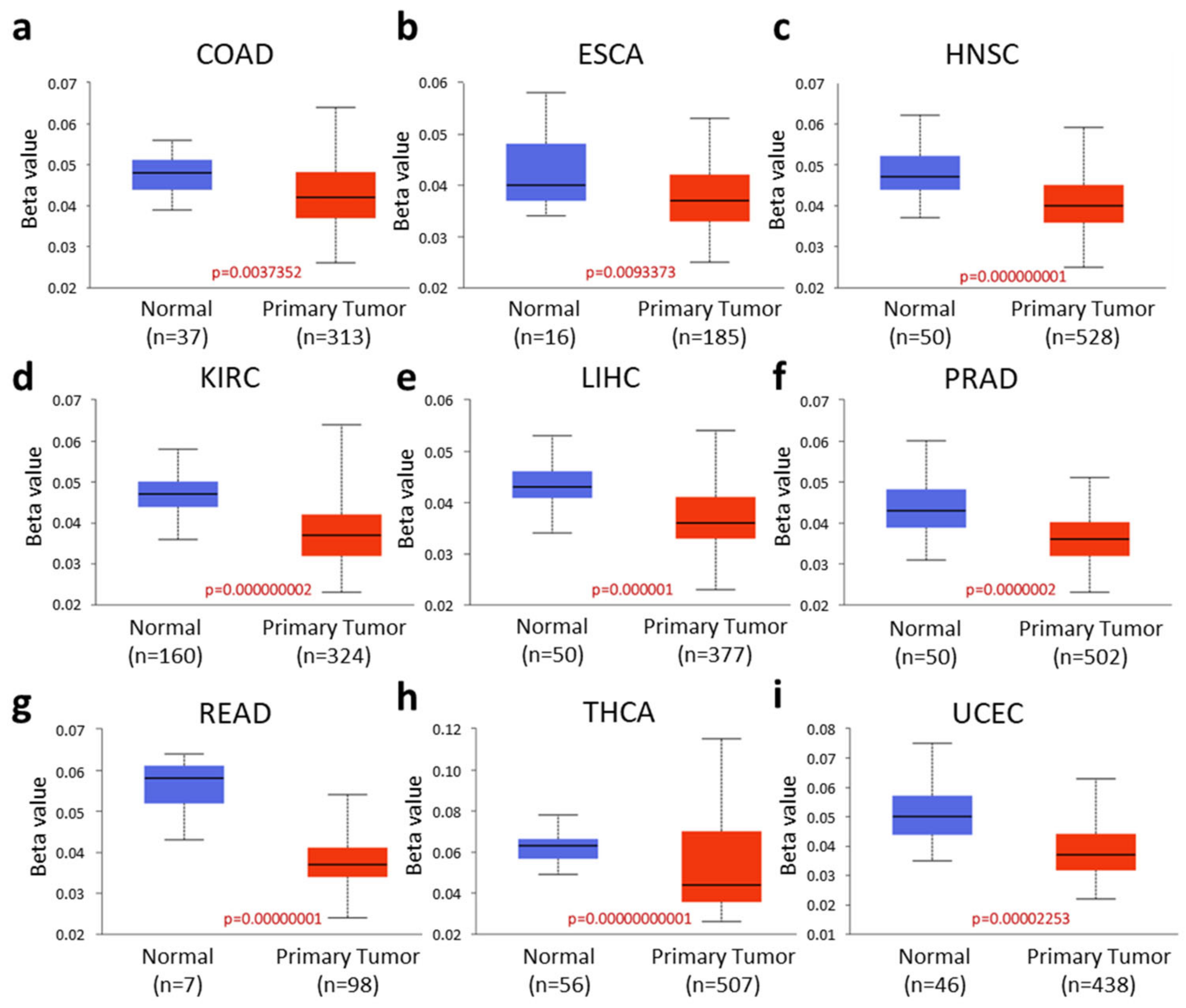
2.2. RASD1 Promoter Methylation Is Low in Many Cancers
2.3. High RASD1 Expression in Tumors Correlates with Longer Survival in KIRC, LGG, and PAAD
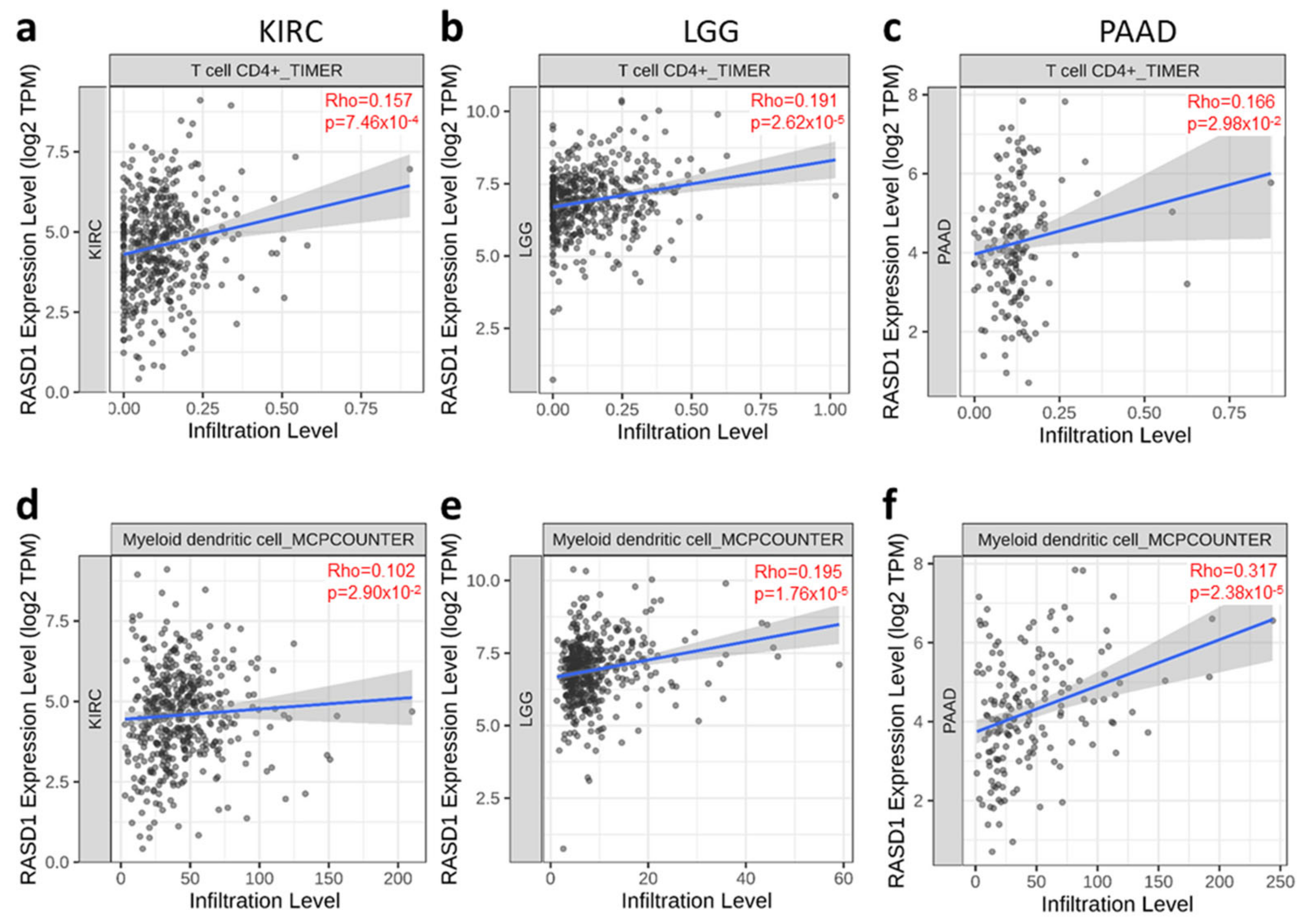
2.4. RASD1 Expression Correlates with the Infiltration of CD4+ T Cells and Myeloid Dendritic Cells in KIRC, LGG, and PAAD
2.5. Enrichment Analysis of Genes with Similar Expression Patterns as RASD1 Suggests a Potential Involvement in Interleukin-4-Mediated Apoptosis and the Transcriptional Regulation of PTEN
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RASD1 | dexamethasone-induced Ras-related protein 1 |
| TCGA | The Cancer Genome Atlas |
| GTEx | Genotype-Tissue Expression |
| PTEN | phosphatase and tensin homolog |
| MAPK | mitogen-activated protein kinase |
| PI3K | phosphoinositide 3-kinase |
| AKT | protein kinase B |
| mTOR | mammalian target of rapamycin |
| THYM | Thymoma |
| UCEC | Uterine corpus endometrial carcinoma |
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder Urothelial Carcinoma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| COAD | Colon adenocarcinoma |
| KICH | Kidney Chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LIHC | Liver hepatocellular carcinoma |
| LUSC | Lung squamous cell carcinoma |
| OV | Ovarian serous cystadenocarcinoma |
| READ | Rectum adenocarcinoma |
| SKCM | Skin Cutaneous Melanoma |
| STAD | Stomach adenocarcinoma |
| ESCA | Esophageal carcinoma |
| HNSC | Head and neck squamous cell carcinoma |
| PRAD | Prostate adenocarcinoma |
| THCA | Thyroid carcinoma |
| MESO | Mesothelioma |
| PAAD | Pancreatic adenocarcinoma |
| LGG | Low grade glioma |
| IL | Interleukin |
| B-ALL | B-cell acute lymphoblastic leukemia |
References
- Soriano, O.; Alcon-Perez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Sutton, M.N.; Lu, Z.; Li, Y.C.; Zhou, Y.; Huang, T.; Reger, A.S.; Hurwitz, A.M.; Palzkill, T.; Logsdon, C.; Liang, X.; et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep. 2019, 29, 3448–3459.e3446. [Google Scholar] [CrossRef]
- Kemppainen, R.J.; Behrend, E.N. Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J. Biol. Chem. 1998, 273, 3129–3131. [Google Scholar] [CrossRef]
- Fang, M.; Jaffrey, S.R.; Sawa, A.; Ye, K.; Luo, X.; Snyder, S.H. Dexras1: A G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron 2000, 28, 183–193. [Google Scholar] [CrossRef]
- Lau, K.F.; Chan, W.M.; Perkinton, M.S.; Tudor, E.L.; Chang, R.C.; Chan, H.Y.; McLoughlin, D.M.; Miller, C.C. Dexras1 interacts with FE65 to regulate FE65-amyloid precursor protein-dependent transcription. J. Biol. Chem. 2008, 283, 34728–34737. [Google Scholar] [CrossRef]
- Cha, J.Y.; Kim, H.J.; Yu, J.H.; Xu, J.; Kim, D.; Paul, B.D.; Choi, H.; Kim, S.; Lee, Y.J.; Ho, G.P.; et al. Dexras1 mediates glucocorticoid-associated adipogenesis and diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 20575–20580. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, G.; Cismowski, M.J.; Wang, G.; Vincent, T.S.; Brown, K.D.; Lanier, S.M. The Ras-related protein AGS1/RASD1 suppresses cell growth. Oncogene 2004, 23, 5858–5863. [Google Scholar] [CrossRef]
- Gao, S.; Jin, L.; Liu, G.; Wang, P.; Sun, Z.; Cao, Y.; Shi, H.; Liu, X.; Shi, Q.; Zhou, X.; et al. Overexpression of RASD1 inhibits glioma cell migration/invasion and inactivates the AKT/mTOR signaling pathway. Sci. Rep. 2017, 7, 3202. [Google Scholar] [CrossRef]
- Tian, J.; Duan, Y.X.; Bei, C.Y.; Chen, J. Calycosin induces apoptosis by upregulation of RASD1 in human breast cancer cells MCF-7. Horm. Metab. Res. 2013, 45, 593–598. [Google Scholar] [CrossRef]
- Liu, X.J.; Li, Y.Q.; Chen, Q.Y.; Xiao, S.J.; Zeng, S.E. Up-regulating of RASD1 and apoptosis of DU-145 human prostate cancer cells induced by formononetin in vitro. Asian Pac. J. Cancer Prev. 2014, 15, 2835–2839. [Google Scholar] [CrossRef]
- Zellinger, B.; Bodenhofer, U.; Englander, I.A.; Kronberger, C.; Strasser, P.; Grambozov, B.; Fastner, G.; Stana, M.; Reitsamer, R.; Sotlar, K.; et al. Hsa-miR-375/RASD1 Signaling May Predict Local Control in Early Breast Cancer. Genes 2020, 11, 1404. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Omberg, L.; Ellrott, K.; Yuan, Y.; Kandoth, C.; Wong, C.; Kellen, M.R.; Friend, S.H.; Stuart, J.; Liang, H.; Margolin, A.A. Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat. Genet. 2013, 45, 1121–1126. [Google Scholar] [CrossRef]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Sadikovic, B.; Al-Romaih, K.; Squire, J.A.; Zielenska, M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr. Genomics 2008, 9, 394–408. [Google Scholar] [CrossRef]
- Chakravarthi, B.V.; Nepal, S.; Varambally, S. Genomic and Epigenomic Alterations in Cancer. Am. J. Pathol. 2016, 186, 1724–1735. [Google Scholar] [CrossRef]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Zhang, S.C.; Hu, Z.Q.; Long, J.H.; Zhu, G.M.; Wang, Y.; Jia, Y.; Zhou, J.; Ouyang, Y.; Zeng, Z. Clinical Implications of Tumor-Infiltrating Immune Cells in Breast Cancer. J. Cancer 2019, 10, 6175–6184. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, M.; Chen, T.; Zhang, B. Characterization of the Immune Cell Infiltration Landscape in Head and Neck Squamous Cell Carcinoma to Aid Immunotherapy. Mol. Ther. Nucleic Acids 2020, 22, 298–309. [Google Scholar] [CrossRef]
- Althubiti, M.A. Mutation Frequencies in Endometrial Cancer Patients of Different Ethnicities and Tumor Grades: An Analytical Study. Saudi J. Med. Med. Sci. 2019, 7, 16–21. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 83. [Google Scholar] [CrossRef]
- Schulz, J.A.; Rodgers, L.T.; Kryscio, R.J.; Hartz, A.M.S.; Bauer, B. Characterization and comparison of human glioblastoma models. BMC Cancer 2022, 22, 844. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Madsen, S.; Peluso, A.A.; Yonamine, C.Y.; Ingerslev, L.R.; Dall, M.; Petersen, P.S.S.; Plucinska, K.; Pradas-Juni, M.; Moreno-Justicia, R.; Gonzalez-Franquesa, A.; et al. Rapid downregulation of DICER is a hallmark of adipose tissue upon high-fat diet feeding. Mol. Cell. Endocrinol. 2025, 595, 112413. [Google Scholar] [CrossRef]
- Ren, M.; Pan, H.; Zhou, X.; Yu, M.; Ji, F. KIAA1429 promotes gastric cancer progression by destabilizing RASD1 mRNA in an m(6)A-YTHDF2-dependent manner. J. Transl. Med. 2024, 22, 584. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Moarii, M.; Boeva, V.; Vert, J.P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genom. 2015, 16, 873. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef]
- Shiah, J.V.; Johnson, D.E.; Grandis, J.R. Transcription Factors and Cancer: Approaches to Targeting. Cancer J. 2023, 29, 38–46. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- de Souza Rocha Simonini, P.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef]
- Chen, Y.; Breeze, C.E.; Zhen, S.; Beck, S.; Teschendorff, A.E. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics Chromatin 2016, 9, 10. [Google Scholar] [CrossRef]
- Herceg, Z.; Hainaut, P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol. Oncol. 2007, 1, 26–41. [Google Scholar] [CrossRef]
- Both, J.; Wu, T.; Bras, J.; Schaap, G.R.; Baas, F.; Hulsebos, T.J. Identification of novel candidate oncogenes in chromosome region 17p11.2-p12 in human osteosarcoma. PLoS ONE 2012, 7, e30907. [Google Scholar] [CrossRef]
- Both, J.; Wu, T.; Ten Asbroek, A.L.; Baas, F.; Hulsebos, T.J. Oncogenic Properties of Candidate Oncogenes in Chromosome Region 17p11.2p12 in Human Osteosarcoma. Cytogenet. Genome Res. 2016, 150, 52–59. [Google Scholar] [CrossRef]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- Miller, M.S.; Miller, L.D. RAS Mutations and Oncogenesis: Not all RAS Mutations are Created Equally. Front. Genet. 2011, 2, 100. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, M.; Novak, L.; Pasquo, A.; Chiaraluce, R.; Turina, P.; Capriotti, E.; Consalvi, V. Analysis and Interpretation of the Impact of Missense Variants in Cancer. Int. J. Mol. Sci. 2021, 22, 5416. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Chen, L.; Grabowski, K.A.; Xin, J.P.; Coleman, J.; Huang, Z.; Espiritu, B.; Alkan, S.; Xie, H.B.; Zhu, Y.; White, F.A.; et al. IL-4 induces differentiation and expansion of Th2 cytokine-producing eosinophils. J. Immunol. 2004, 172, 2059–2066. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Ohara, J.; Myers, C.D.; Layton, J.E.; Krammer, P.H.; Paul, W.E. Serological, biochemical, and functional identity of B cell-stimulatory factor 1 and B cell differentiation factor for IgG1. J. Exp. Med. 1985, 162, 1726–1731. [Google Scholar] [CrossRef]
- Coffman, R.L.; Ohara, J.; Bond, M.W.; Carty, J.; Zlotnik, A.; Paul, W.E. B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J. Immunol. 1986, 136, 4538–4541. [Google Scholar] [CrossRef]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef]
- Salvatore, L.; Calegari, M.A.; Loupakis, F.; Fassan, M.; Di Stefano, B.; Bensi, M.; Bria, E.; Tortora, G. PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target. Cancers 2019, 11, 1765. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Wang, W.; Hao, Q.; Liu, Y. High RASD1 transcript levels at diagnosis predicted poor survival in adult B-cell acute lymphoblastic leukemia patients. Leuk. Res. 2019, 80, 26–32. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug. Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer. 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene ID | PCC 1 |
|---|---|---|
| DLG5-AS1 | ENSG00000233871.2 | 0.57 |
| TMOD1 | ENSG00000136842.13 | 0.56 |
| SCARA3 | ENSG00000168077.13 | 0.54 |
| NLRP1 | ENSG00000091592.15 | 0.53 |
| PDE8B | ENSG00000113231.13 | 0.53 |
| SMG6 | ENSG00000070366.13 | 0.53 |
| DST | ENSG00000151914.17 | 0.53 |
| CADPS | ENSG00000163618.17 | 0.52 |
| TMEM132E | ENSG00000181291.6 | 0.52 |
| KCNH1-IT1 | ENSG00000234233.1 | 0.52 |
| HMBOX1 | ENSG00000147421.17 | 0.51 |
| FJX1 | ENSG00000179431.6 | 0.51 |
| FRMPD3 | ENSG00000147234.10 | 0.51 |
| RING1 | ENSG00000204227.4 | 0.51 |
| MED9 | ENSG00000141026.5 | 0.51 |
| TPST1 | ENSG00000169902.13 | 0.5 |
| SPEG | ENSG00000072195.14 | 0.5 |
| ROM1 | ENSG00000149489.8 | 0.49 |
| C1QL1 | ENSG00000131094.3 | 0.49 |
| TCEAL3 | ENSG00000196507.10 | 0.49 |
| ATN1 | ENSG00000111676.14 | 0.49 |
| PRAF2 | ENSG00000243279.3 | 0.49 |
| NAV2 | ENSG00000166833.19 | 0.49 |
| C6orf1 | ENSG00000186577.11 | 0.49 |
| DCAKD | ENSG00000172992.11 | 0.49 |
| ST8SIA1 | ENSG00000111728.10 | 0.48 |
| LRSAM1 | ENSG00000148356.13 | 0.48 |
| GALNT15 | ENSG00000131386.17 | 0.48 |
| SDR39U1 | ENSG00000100445.16 | 0.48 |
| JAKMIP2 | ENSG00000176049.15 | 0.48 |
| RP11-209D14.2 | ENSG00000261033.1 | 0.48 |
| ABCA3 | ENSG00000167972.13 | 0.48 |
| GAP43 | ENSG00000172020.12 | 0.47 |
| TCEA2 | ENSG00000171703.16 | 0.47 |
| TMEM200C | ENSG00000206432.4 | 0.47 |
| SEC14L2 | ENSG00000100003.17 | 0.47 |
| ID4 | ENSG00000172201.10 | 0.47 |
| BAALC | ENSG00000164929.16 | 0.47 |
| USP20 | ENSG00000136878.12 | 0.47 |
| PMP2 | ENSG00000147588.6 | 0.47 |
| ADD1 | ENSG00000087274.16 | 0.47 |
| BCL6 | ENSG00000113916.17 | 0.47 |
| FAM127A | ENSG00000134590.13 | 0.47 |
| FAM167A | ENSG00000154319.14 | 0.47 |
| BBS2 | ENSG00000125124.11 | 0.47 |
| FAM69B | ENSG00000165716.9 | 0.47 |
| GFAP | ENSG00000131095.11 | 0.47 |
| SPOCK2 | ENSG00000107742.12 | 0.47 |
| RALGDS | ENSG00000160271.14 | 0.46 |
| BAALC-AS2 | ENSG00000236939.2 | 0.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsueh, H.-Y.; Gumpper-Fedus, K.; Poelstra, J.W.; Pitter, K.L.; Cruz-Monserrate, Z. Pan-Cancer Analysis Identifies a Ras-Related GTPase as a Potential Modulator of Cancer. Int. J. Mol. Sci. 2025, 26, 4419. https://doi.org/10.3390/ijms26094419
Hsueh H-Y, Gumpper-Fedus K, Poelstra JW, Pitter KL, Cruz-Monserrate Z. Pan-Cancer Analysis Identifies a Ras-Related GTPase as a Potential Modulator of Cancer. International Journal of Molecular Sciences. 2025; 26(9):4419. https://doi.org/10.3390/ijms26094419
Chicago/Turabian StyleHsueh, Hsiang-Yin, Kristyn Gumpper-Fedus, Jelmer W. Poelstra, Kenneth L. Pitter, and Zobeida Cruz-Monserrate. 2025. "Pan-Cancer Analysis Identifies a Ras-Related GTPase as a Potential Modulator of Cancer" International Journal of Molecular Sciences 26, no. 9: 4419. https://doi.org/10.3390/ijms26094419
APA StyleHsueh, H.-Y., Gumpper-Fedus, K., Poelstra, J. W., Pitter, K. L., & Cruz-Monserrate, Z. (2025). Pan-Cancer Analysis Identifies a Ras-Related GTPase as a Potential Modulator of Cancer. International Journal of Molecular Sciences, 26(9), 4419. https://doi.org/10.3390/ijms26094419