
Comparative Chloroplast Genomics of Actinidia deliciosa Cultivars: Insights into Positive Selection and Population Evolution

Abstract
1. Introduction
2. Results
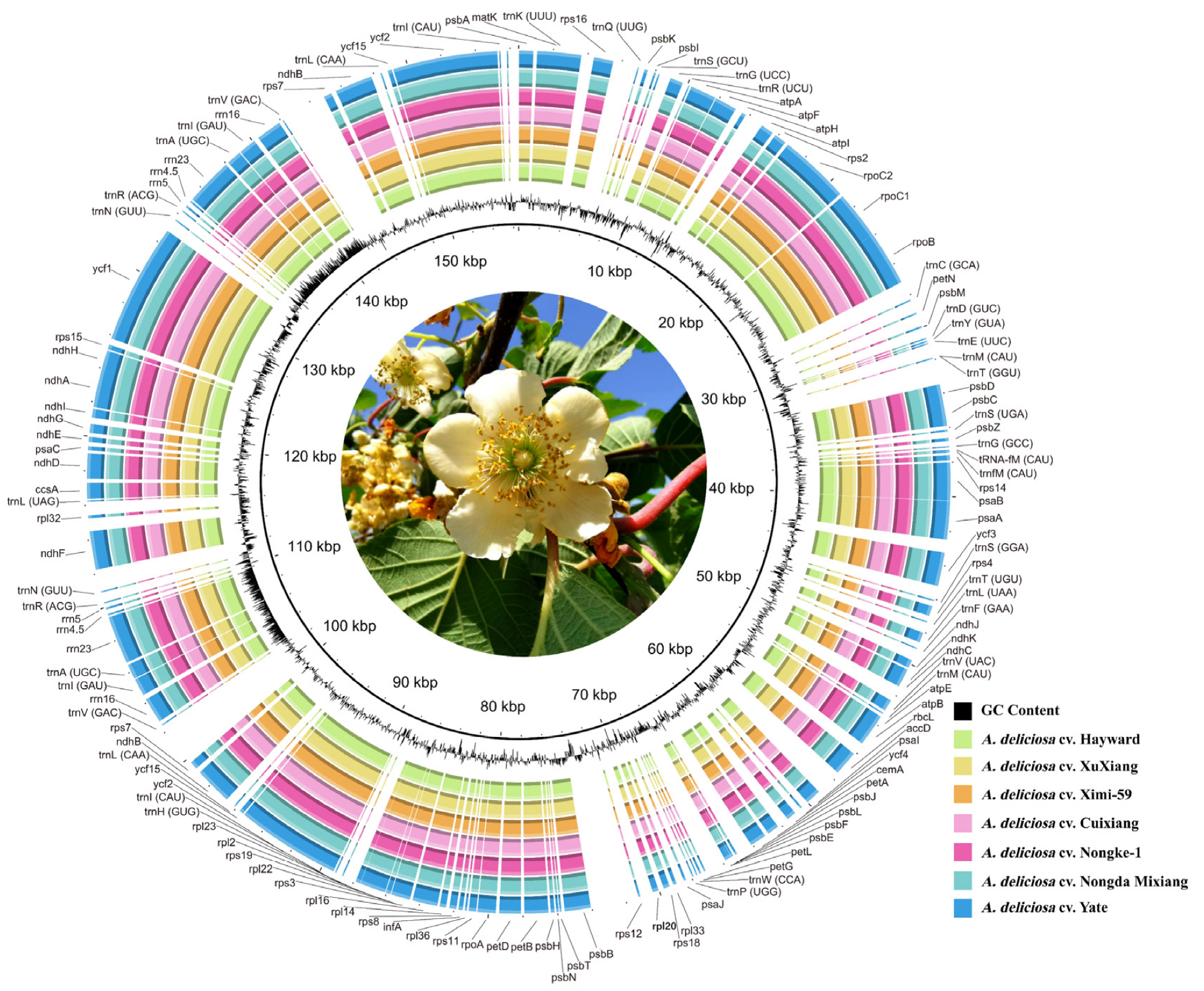
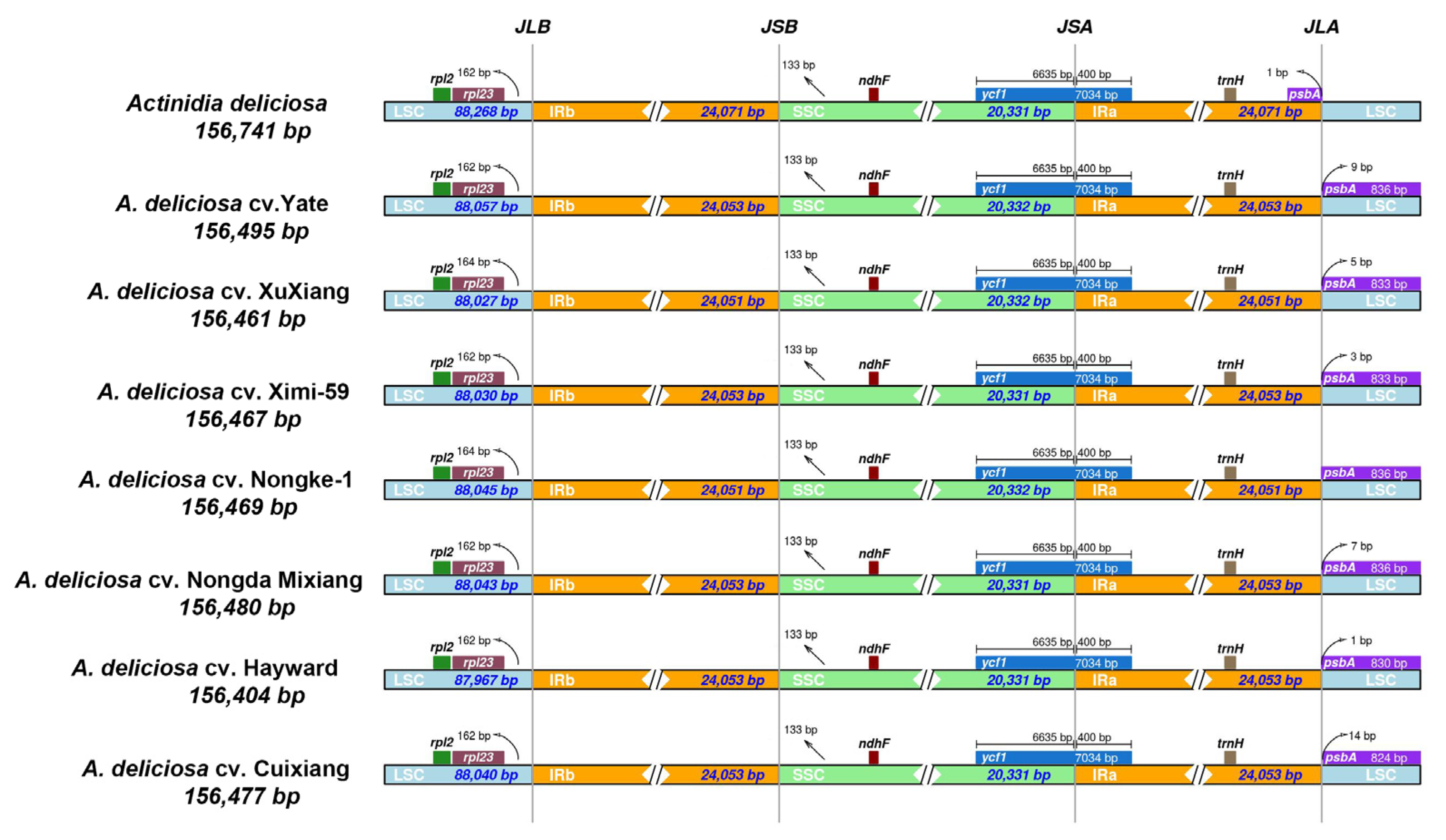
2.1. Chloroplast Genome Features of A. deliciosa Cultivars
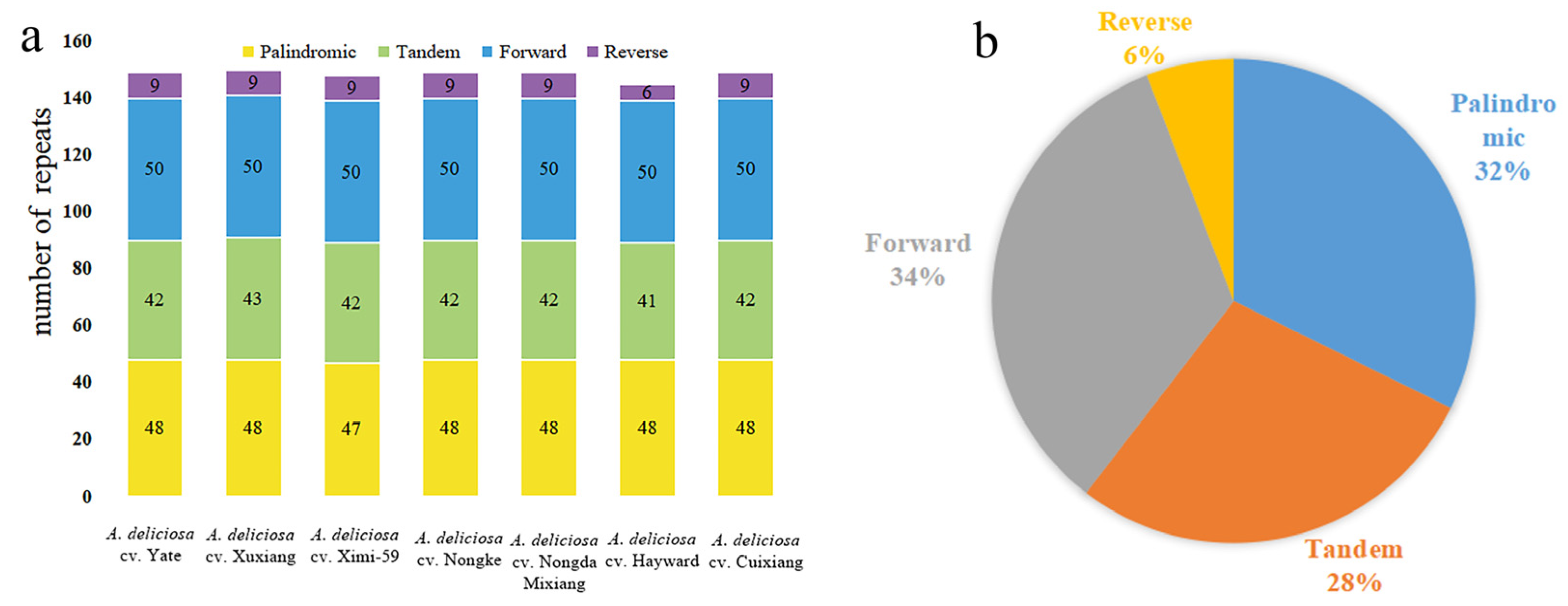
2.2. Distribution and Characterization of Repeat Elements
2.3. Sequence Divergence Patterns of Chloroplast Genomes
2.4. Positive Selection Genes
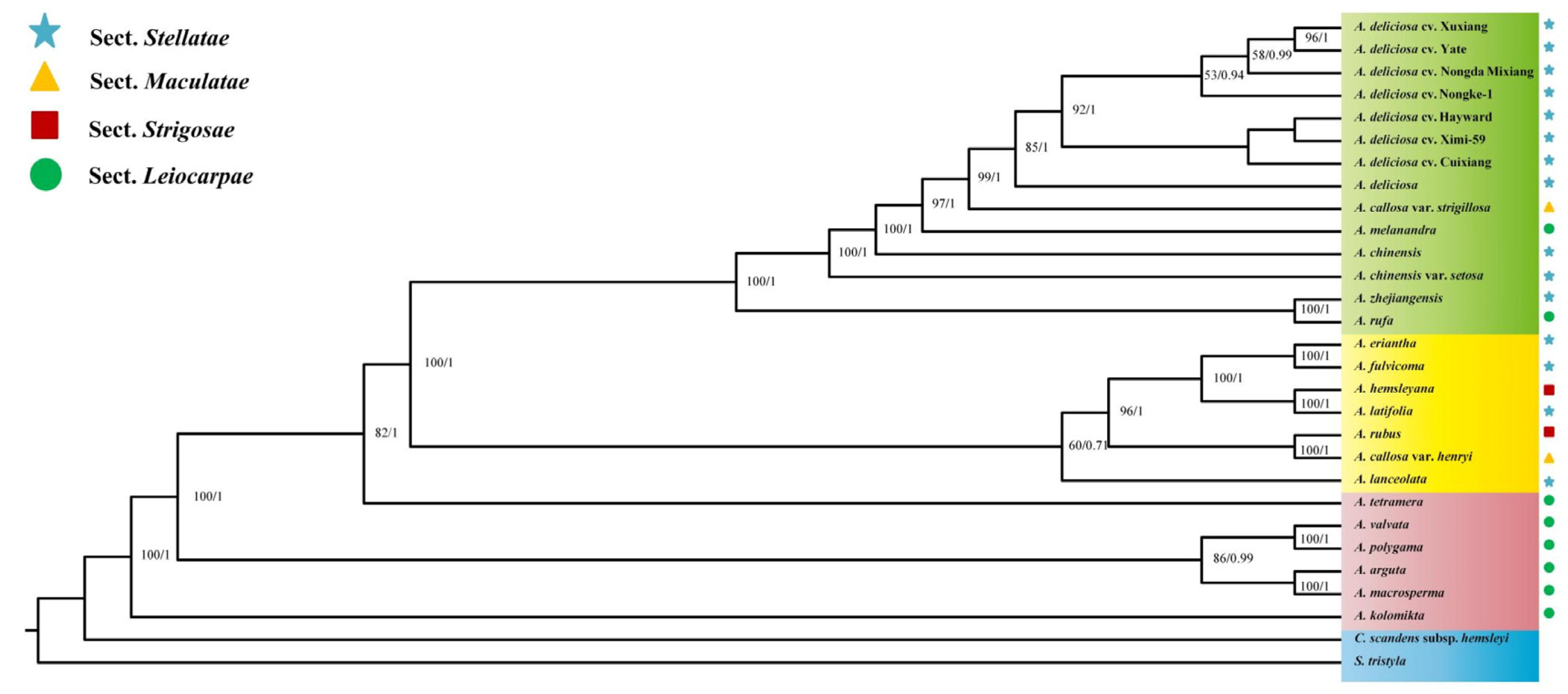
2.5. Genetic Evolutionary Analysis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plant Material and DNA Extraction
5.2. Chloroplast Genomes Assembly and Annotation
5.3. Repeat Elements Analysis
5.4. Comparative Chloroplast Genome Analysis
5.5. Positive Selection Analysis
5.6. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, C.F.; Chen, Y.C.; Wang, Y.S. Actinidiaceae. Flora Reipublicae Pop. Sin. 1984, 49, 2. [Google Scholar]
- Li, W.C.; Wang, K.L.; Peng, Y.J.; Zhou, Y.; Liu, Y.B. Distribution patterns and driving factors of mixed-ploidy Actinidia species in China. Sci. Hortic. 2024, 331, 113149. [Google Scholar] [CrossRef]
- Huang, S.X.; Ding, J.; Deng, D.J.; Tang, W.; Sun, H.; Liu, D.Y.; Zhang, L.; Niu, X.L.; Zhang, X.; Meng, M.; et al. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 2013, 4, 2640. [Google Scholar] [CrossRef] [PubMed]
- Perez-Burillo, S.; Oliveras, M.J.; Quesada, J.; Rufian-Henares, J.A.; Pastoriza, S. Relationship between composition and bioactivity of persimmon and kiwifruit. Food Res. Int. 2018, 105, 461–472. [Google Scholar] [CrossRef]
- Ferguson, A.R.; Huang, H. Genetic resources of kiwifruit: Domestication and breeding. Hortic. Rev. 2007, 33, 1–121. [Google Scholar]
- Huang, H.; Ferguson, A.R. Review: Kiwifruit in China. N. Z. J. Crop Hortic. Sci. 2001, 29, 1–14. [Google Scholar] [CrossRef]
- Dunn, S.T. A revision of the genus Actinidia, Lindl. Bot. J. Linn. Soc. 1911, 39, 394–410. [Google Scholar] [CrossRef]
- Li, H.L. A taxonomic review of the genus Actinidia. J. Arnold Arbor. 1952, 33, 1–61. [Google Scholar] [CrossRef]
- Cipriani, G.; Testolin, R.; Gardner, R. Restriction-site variation of PCR-amplified chloroplast DNA regions and its implication for the evolution and taxonomy of Actinidia. Theor. Appl. Genet. 1998, 96, 389–396. [Google Scholar] [CrossRef]
- Chat, J.; Jáuregui, B.; Petit, R.J.; Nadot, S. Reticulate evolution in kiwifruit (Actinidia, Actinidiaceae) identified by comparing their maternal and paternal phylogenies. Am. J. Bot. 2004, 91, 736–747. [Google Scholar] [CrossRef]
- Yao, X.H.; Wang, S.B.; Wang, Z.P.; Li, D.W.; Jiang, Q.; Zhang, Q.; Zhong, C.H.; Huang, H.W.; Liu, Y.F. The genome sequencing and comparative analysis of a wild kiwifruit Actinidia eriantha. Mol. Hortic. 2022, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Q.; Xie, X.D.; Yang, M.J.; Atak, A.; Zhong, C.H.; Dawei, L. Characterization of the complete chloroplast genome of Actinidia hemsleyana. Mitochondrial DNA Part B 2021, 6, 3259–3260. [Google Scholar]
- Liu, H.; Liu, X.; Sun, C.; Li, H.L.; Li, Z.X.; Guo, Y.; Fu, X.Q.; Liao, Q.H.; Zhang, W.L.; Liu, Y.Q. Chloroplast genome comparison and phylogenetic analysis of the commercial variety Actinidia chinensis ‘Hongyang’. Genes 2023, 14, 2136. [Google Scholar] [CrossRef]
- Lin, Q.H.; Hu, S.Q.; Wu, Z.H.; Huang, Y.H.; Wang, S.; Shi, W.B.; Zhu, B.Y. Comparative chloroplast genomics provides insights into the phylogenetic relationships and evolutionary history for Actinidia species. Sci. Rep. 2025, 15, 13291. [Google Scholar] [CrossRef]
- Wang, L.C.; Liu, B.; Yang, Y.R.; Zhuang, Q.G.; Chen, S.J.; Liu, Y.S.; Huang, S.X. The comparative studies of complete chloroplast genomes in Actinidia (Actinidiaceae): Novel insights into heterogenous variation, clpP gene annotation and phylogenetic relationships. Mol. Genet. Genom. 2022, 297, 535–551. [Google Scholar] [CrossRef]
- Liu, X.; Sun, C.; Li, M.Z.; Liu, J.; Zhang, W.L.; Liu, R.; Liu, Y.Q.; Le, Y.Z.; Lan, J.B. The complete chloroplast genome sequence of Actinidia chinensis Planch.‘Hongyang’, a typical red core pulp in China. Mitochondrial DNA Part B 2022, 7, 593–595. [Google Scholar] [CrossRef]
- Azarin, K.; Usatov, A.; Makarenko, M.; Kozel, N.; Kovalevich, A.; Dremuk, I.; Yemelyanova, A.; Logacheva, M.; Fedorenko, A.; Averina, N. A point mutation in the photosystem I P700 chlorophyll a apoprotein A1 gene confers variegation in Helianthus annuus L. Plant Mol. Biol. 2020, 103, 373–389. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Yao, X.H.; Tang, P.; Li, Z.Z.; Li, D.W.; Liu, Y.F.; Huang, H.W. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. Public Libr. Sci. ONE 2015, 10, e0129347. [Google Scholar] [CrossRef]
- Bock, R. Structure, function, and inheritance of plastid genomes. In Cell and Molecular Biology of Plastids; Springer: Berlin/Heidelberg, Germany, 2007; pp. 29–63. [Google Scholar]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Abdullah; Mehmood, F.; Shahzadi, I.; Ali, Z.; Islam, M.; Naeem, M.; Mirza, B.; Lockhart, P.J.; Ahmed, I.; Waheed, M.T. Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. 2021, 59, 388–402. [Google Scholar] [CrossRef]
- Lopes, A.d.S.; Pacheco, T.G.; Silva, O.N.d.; Vieira, L.d.N.; Guerra, M.P.; Mattar, E.P.L.; Baura, V.A.d.; Balsanelli, E.; Souza, E.M.d.; Pedrosa, F.d.O.; et al. Plastid genome evolution in Amazonian açaí palm (Euterpe oleracea Mart.) and Atlantic forest açaí palm (Euterpe edulis Mart.). Plant Mol. Biol. 2021, 105, 559–574. [Google Scholar] [CrossRef]
- Ahmed, I.; Biggs, P.J.; Matthews, P.J.; Collins, L.J.; Hendy, M.D.; Lockhart, P.J. Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 2012, 4, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.J.; Wang, T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.H.; Zhang, J.J.; Tian, H.; Yao, X.H. Characterization of 39 novel EST-SSR markers for Liriodendron tulipifera and cross-species amplification in L. chinense (Magnoliaceae). Am. J. Bot. 2012, 99, e460–e464. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.R.; Huang, P.; Guo, W.Y.; Zheng, Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 2020, 53, 21. [Google Scholar] [CrossRef]
- Flickinger, R. Polymorphism of simple sequence repeats may quantitatively regulate gene transcription. Exp. Cell Res. 2020, 390, 111969. [Google Scholar] [CrossRef]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). Public Libr. Sci. ONE 2012, 7, e36869. [Google Scholar] [CrossRef]
- Wolfson, R.; Higgins, K.G.; Sears, B.B. Evidence for replication slippage in the evolution of Oenothera chloroplast DNA. Mol. Biol. Evol. 1991, 8, 709–720. [Google Scholar]
- Brázda, V.; Lýsek, J.; Bartas, M.; Fojta, M. Complex analyses of short inverted repeats in all sequenced chloroplast DNAs. BioMed Res. Int. 2018, 2018, 1097018. [Google Scholar] [CrossRef]
- Korkovelos, A.E.; Mavromatis, A.G.; Huang, W.G.; Hagidimitriou, M.; Giakoundis, A.; Goulas, C.K. Effectiveness of SSR molecular markers in evaluating the phylogenetic relationships among eight Actinidia species. Sci. Hortic. 2008, 116, 305–310. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 4302–4315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (poaceae: Bambusoideae). Public Libr. Sci. ONE 2011, 6, e20596. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef]
- Oldenburg, D.J.; Bendich, A.J. Most chloroplast DNA of maize seedlings in linear molecules with defined ends and branched forms. J. Mol. Biol. 2004, 335, 953–970. [Google Scholar] [CrossRef]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef]
- Wang, N.; Chen, S.F.; Xie, L.; Wang, L.; Feng, Y.Y.; Lv, T.; Fang, Y.M.; Ding, H. The complete chloroplast genomes of three Hamamelidaceae species: Comparative and phylogenetic analyses. Ecol. Evol. 2022, 12, e8637. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Yan, M.; Ding, Y.; Huo, Y.; Yuan, Z.H. Characterization and comparative analysis of the complete chloroplast genome sequence from Prunus avium ‘Summit’. Plant Biol. 2019, 7, e8210. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.Y.; Gao, H.H.; Zhu, Y.J.; Xu, J.; Pang, X.H.; Yao, H.; Sun, C.; Li, X.E.; Li, C.Y.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. Public Libr. Sci. ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Ghulam, M.M.; Zghidi-Abouzid, O.; Lambert, E.; Lerbs-Mache, S.; Merendino, L. Transcriptional organization of the large and the small ATP synthase operons, atpI/H/F/A and atpB/E, in Arabidopsis thaliana chloroplasts. Plant Mol. Biol. 2012, 79, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Chi, W.; Ouyang, M.; He, B.Y.; Chen, F.; Zhang, L.X. PAB is an assembly chaperone that functions downstream of chaperonin 60 in the assembly of chloroplast ATP synthase coupling factor 1. Proc. Natl. Acad. Sci. USA 2015, 112, 4152–4157. [Google Scholar] [CrossRef]
- Tiller, N.; Bock, R. The translational apparatus of plastids and its role in plant development. Mol. Plant 2014, 7, 1105–1120. [Google Scholar] [CrossRef]
- Rosado, A.; Li, R.; van de Ven, W.; Hsu, E.; Raikhel, N.V. Arabidopsis ribosomal proteins control developmental programs through translational regulation of auxin response factors. Proc. Natl. Acad. Sci. USA 2012, 109, 19537–19544. [Google Scholar] [CrossRef]
- Allahverdiyeva, Y.; Mamedov, F.; Mäenpää, P.; Vass, I.; Aro, E.M. Modulation of photosynthetic electron transport in the absence of terminal electron acceptors: Characterization of the rbcL deletion mutant of tobacco. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1709, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, M.V.; Filatov, D.A. Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol. Biol. 2007, 7, 73–82. [Google Scholar] [CrossRef]
- Piot, A.; Hackel, J.; Christin, P.A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef]
- Drescher, A.; Ruf, S.; Calsa, T.J.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Sun, G.L.; Zhang, D.M. Molecular evolution and phylogeny of the angiosperm ycf2 gene. J. Mol. Biol. 2010, 48, 240–248. [Google Scholar] [CrossRef]
- Golding, G.B.; Dean, A.M. The structural basis of molecular adaptation. Mol. Biol. Evol. 1998, 15, 355–369. [Google Scholar] [CrossRef]
- Abdullah; Henriquez, C.L.; Mehmood, F.; Carlsen, M.M.; Islam, M.; Waheed, M.T.; Pocza, P.; Croat, T.B.; Ahmed, I. Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): Unique inverted repeat expansion and contraction affect rate of evolution. J. Mol. Evol. 2020, 88, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.F.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Rife, M.B.; Dave, B.; Anto, N.; et al. HyPhy 2.5—A customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.H.; Liu, L.; Yan, M.K.; Li, D.W.; Zhong, C.H.; Huang, H.W. Exon primed intron-crossing (EPIC) markers reveal natural hybridization and introgression in Actinidia (Actinidiaceae) with sympatric distribution. Syst. Ecol. 2015, 59, 246–255. [Google Scholar] [CrossRef]
- He, Z.C.; Zhang, X.Y.; Zhong, Y.; Ye, L. Phylogenetic relationships of Actinidia and related genera based on micromorphological characters of foliar trichomes. Genet. Resour. Crop Evol. 2000, 47, 12. [Google Scholar] [CrossRef]
- Li, J.Q.; Huang, H.W.; Sang, T. Molecular phylogeny and infrageneric classification of Actinidia (Actinidiaceae). Syst. Bot. 2002, 27, 408–415. [Google Scholar]
- Testolin, R.; Ferguson, A.R. Isozyme polymorphism in the genus Actinidia and the origin of the kiwifruit genome. Syst. Bot. 1997, 22, 685–700. [Google Scholar] [CrossRef]
- Li, J.L.; Wang, S.; Yu, J.; Wang, L.; Zhou, S.L. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. Public Libr. Sci. 2012, 7, e30619. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Shi, L.C.; Chen, H.M.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Frazer, K.A.; Lior, P.; Alexander, P.; Rubin, E.M.; Inna, D. Vista: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. Irscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Hiratsuka, J.; Shimada, H.; Whittier, R.; Ishibashi, T.; Sakamoto, M.; Mori, M.; Kondo, C.; Honji, Y.; Sun, C.R.; Meng, B.Y.; et al. The complete sequence of the rice (Oryza sativa) chloroplast genome: Intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 1989, 217, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Shahid Masood, M.; Nishikawa, T.; Fukuoka, S.; Njenga, P.K.; Tsudzuki, T.; Kadowaki, K. The complete nucleotide sequence of wild rice (Oryza nivara) chloroplast genome: First genome wide comparative sequence analysis of wild and cultivated rice. Gene 2004, 340, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | A. deliciosa cv. Cuixiang | A. deliciosa cv. XuXiang | A. deliciosa cv. Hayward | A. deliciosa cv. Nongda Mixiang | A. deliciosa cv. Yate | A. deliciosa cv. Nongke-1 | A. deliciosa cv. Ximi-59 |
|---|---|---|---|---|---|---|---|
| Size (bp) | 156,477 | 156,461 | 156,404 | 156,480 | 156,495 | 156,479 | 156,467 |
| LSC length (bp) | 88,040 | 88,027 | 87,967 | 88,043 | 88,057 | 88,045 | 88,030 |
| SSC length (bp) | 20,331 | 20,332 | 20,331 | 20,331 | 20,332 | 20,332 | 20,331 |
| IR length (bp) | 48,106 | 48,102 | 48,106 | 48,106 | 48,106 | 48,102 | 48,106 |
| Coding regions (bp) | 76,941 | 76,939 | 76,395 | 76,941 | 76,941 | 76,939 | 79,941 |
| Non-coding regions (bp) | 79,536 | 79,522 | 80,009 | 79,539 | 79,554 | 79,540 | 76,526 |
| Number of genes | 131 | 131 | 131 | 131 | 131 | 131 | 131 |
| Protein-coding genes | 83 | 83 | 83 | 83 | 83 | 83 | 83 |
| tRNA genes | 40 | 40 | 40 | 40 | 40 | 40 | 40 |
| rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| GC content (%) | 37.20 | 37.20 | 37.20 | 37.20 | 37.20 | 37.20 | 37.20 |
| GC content of LSC (%) | 35.50 | 35.50 | 35.50 | 35.50 | 35.50 | 35.50 | 35.50 |
| GC content of SSC (%) | 31.10 | 31.10 | 31.10 | 31.10 | 31.10 | 31.10 | 31.10 |
| GC content of IR (%) | 42.90 | 42.90 | 42.90 | 42.90 | 42.90 | 42.90 | 42.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Yang, Y.; Zhang, X.; Zhao, W.; Zhang, Q.; Luo, C.; Xie, Y.; Li, Z.; Wang, X. Comparative Chloroplast Genomics of Actinidia deliciosa Cultivars: Insights into Positive Selection and Population Evolution. Int. J. Mol. Sci. 2025, 26, 4387. https://doi.org/10.3390/ijms26094387
He X, Yang Y, Zhang X, Zhao W, Zhang Q, Luo C, Xie Y, Li Z, Wang X. Comparative Chloroplast Genomics of Actinidia deliciosa Cultivars: Insights into Positive Selection and Population Evolution. International Journal of Molecular Sciences. 2025; 26(9):4387. https://doi.org/10.3390/ijms26094387
Chicago/Turabian StyleHe, Xiaojing, Yang Yang, Xingya Zhang, Weimin Zhao, Qijing Zhang, Caiyun Luo, Yanze Xie, Zhonghu Li, and Xiaojuan Wang. 2025. "Comparative Chloroplast Genomics of Actinidia deliciosa Cultivars: Insights into Positive Selection and Population Evolution" International Journal of Molecular Sciences 26, no. 9: 4387. https://doi.org/10.3390/ijms26094387
APA StyleHe, X., Yang, Y., Zhang, X., Zhao, W., Zhang, Q., Luo, C., Xie, Y., Li, Z., & Wang, X. (2025). Comparative Chloroplast Genomics of Actinidia deliciosa Cultivars: Insights into Positive Selection and Population Evolution. International Journal of Molecular Sciences, 26(9), 4387. https://doi.org/10.3390/ijms26094387