
Integrated Transcriptomic and Epigenomic Analysis Reveals Mechanisms Underlying Melanotic Spot Formation in Red Tilapia (Oreochromis spp.)

 ,
,  and
and
Abstract
1. Introduction
2. Results
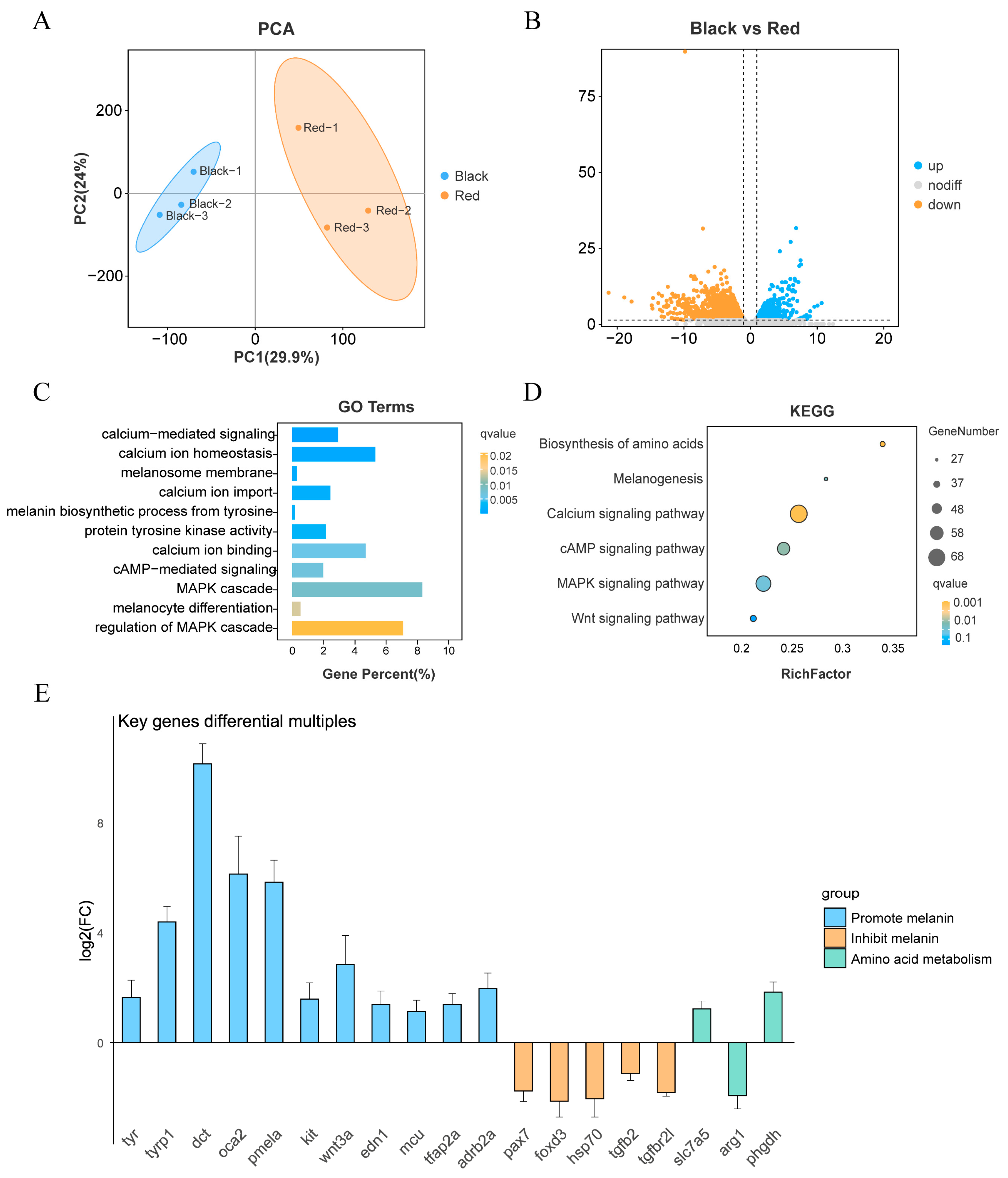
2.1. Transcriptome Data Processing and Alignment
2.2. Transcriptome Differential Expression and Enrichment Analysis
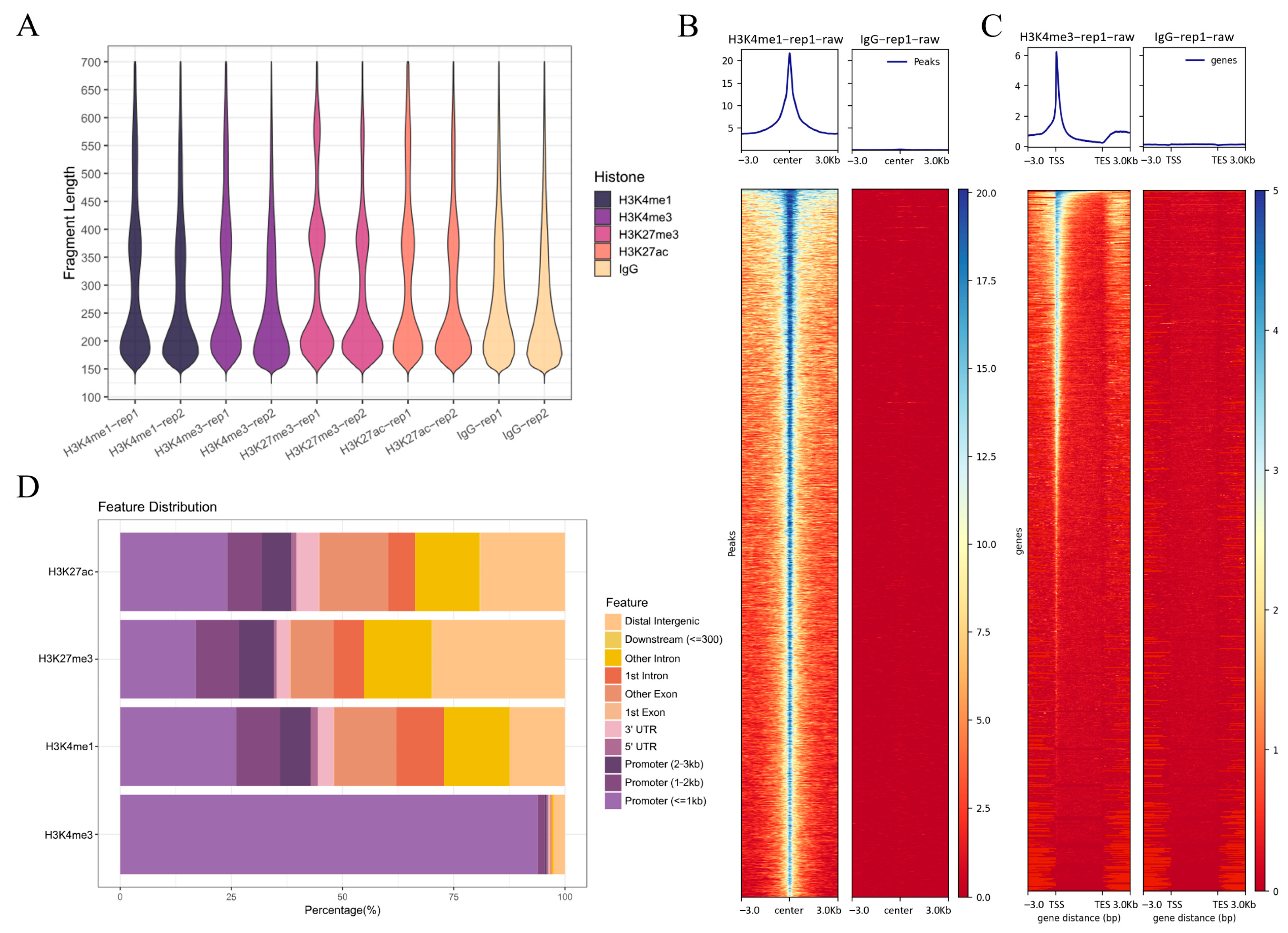
2.3. CUT&Tag Data Processing and Alignment
2.4. Distribution and Enrichment Characteristics of Histone Modification Signals in the Skin Tissue of Black Spot and Red Regions
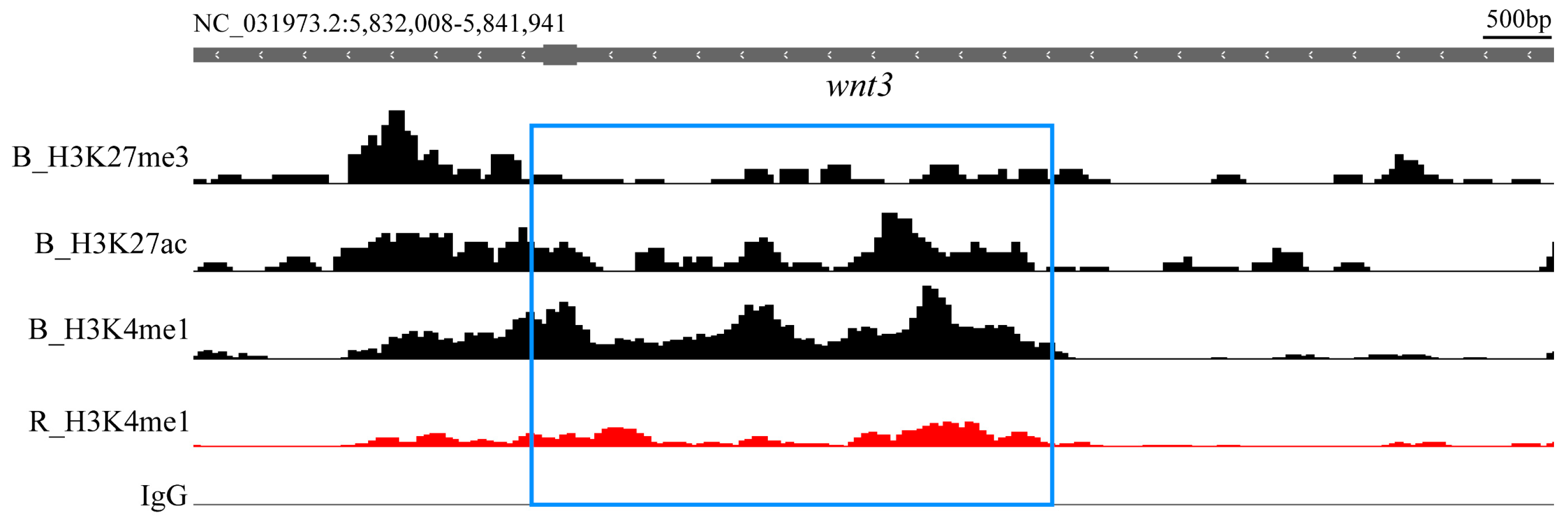
2.5. Comparison and Analysis of Differential Histone Modification Signals
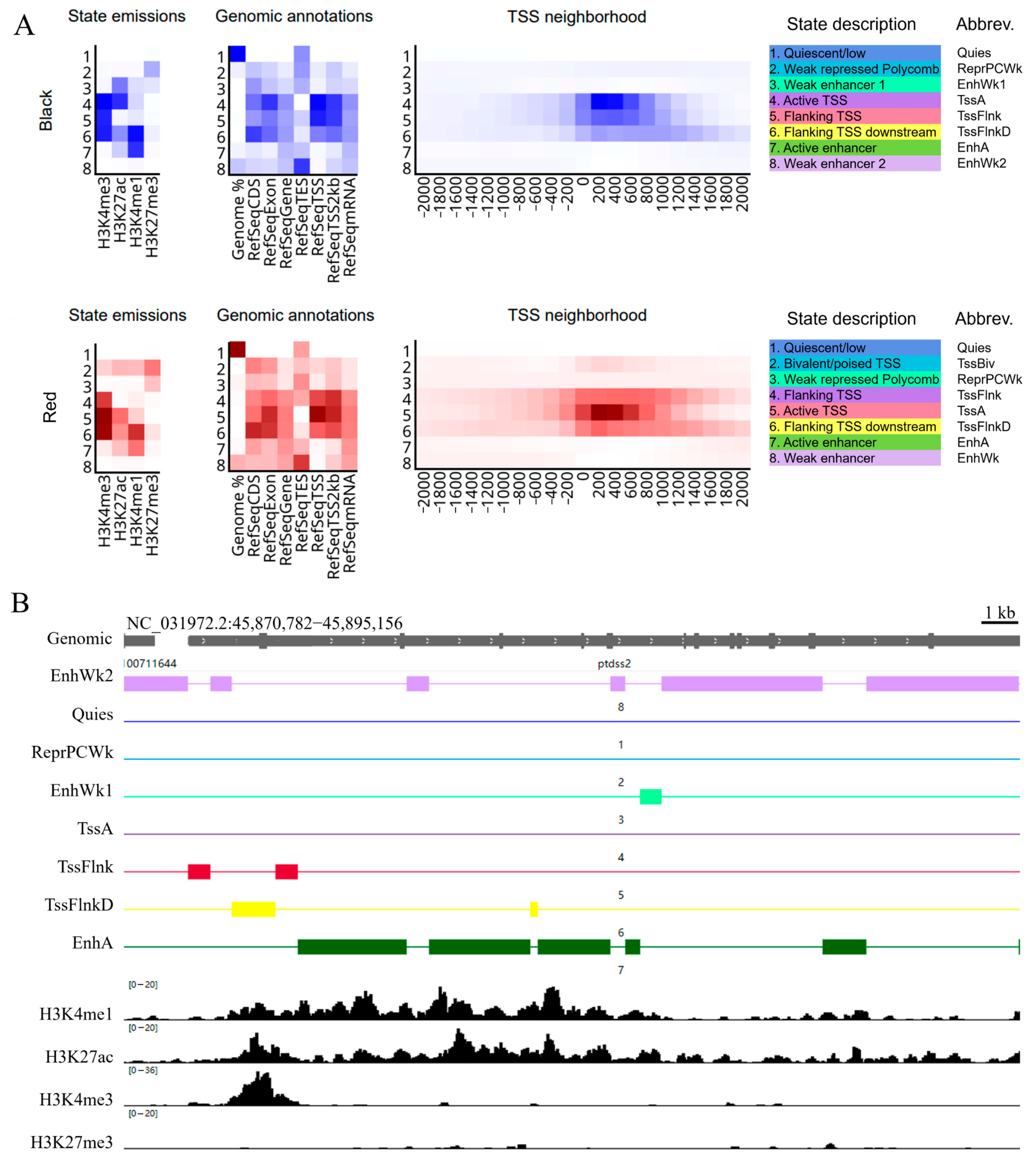
2.6. Chromatin State Analysis of the Black Spot and Red Regions
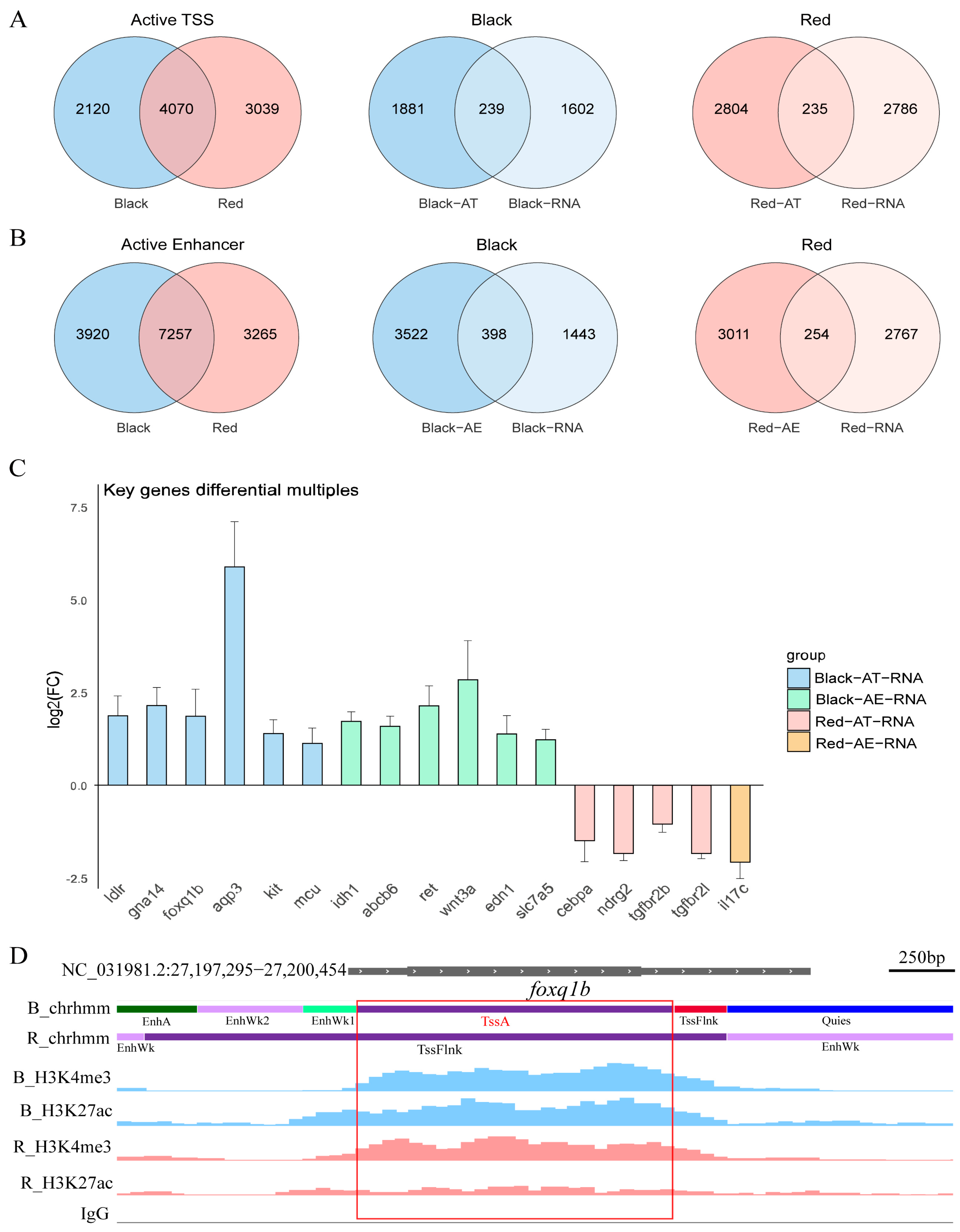
2.7. Chromatin State and Transcriptome Integrated Analysis
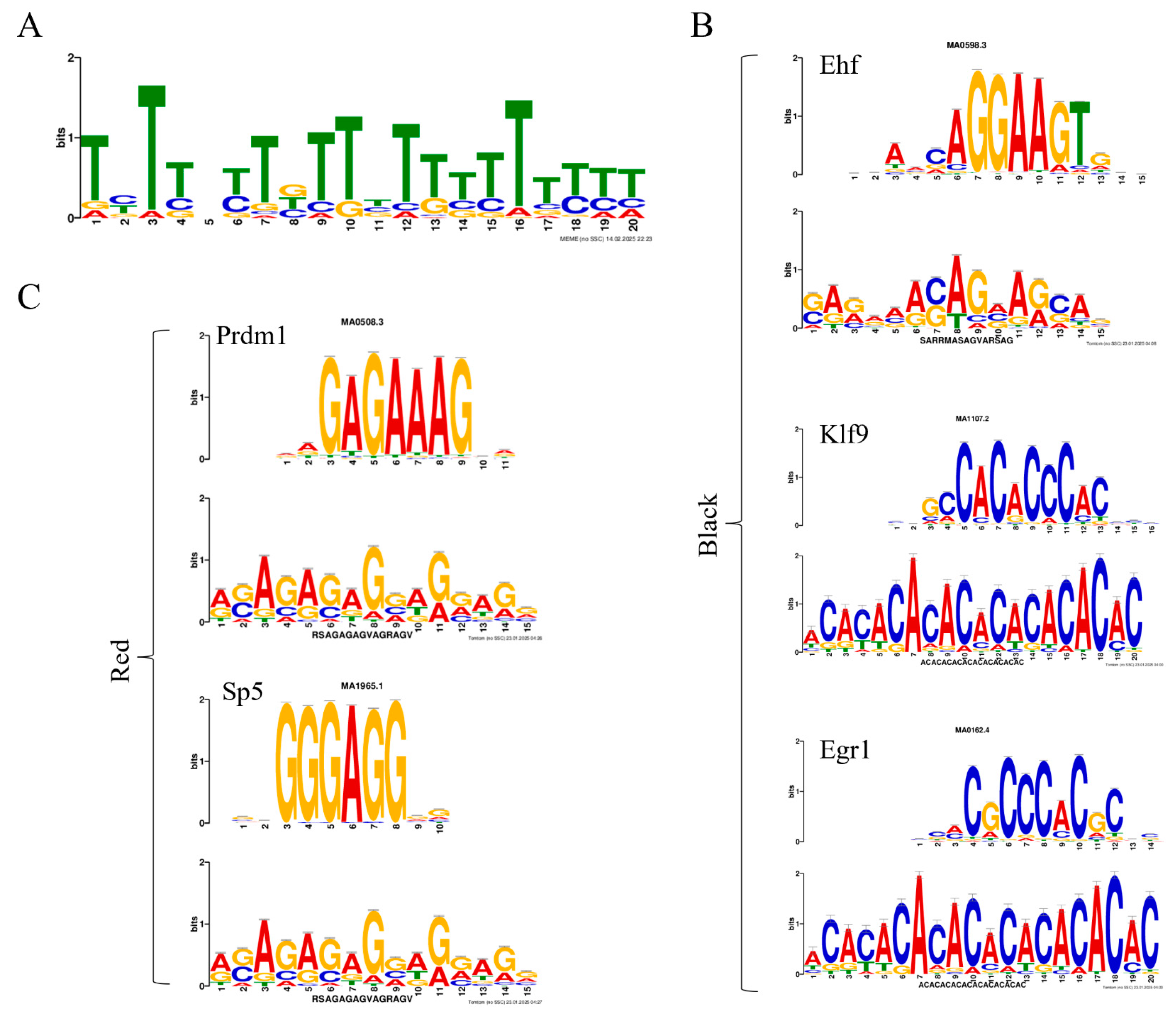
2.8. Motif and Transcription Factor Prediction
3. Discussion
3.1. Gene Regulatory Network and the Molecular Mechanism of Spot Formation
3.2. Histone Modification Screening of Key Regulatory Elements
3.3. Epigenetic and Transcriptional Regulatory Networks of Pigment Differences Between the Black Spot and Red Regions in Red Tilapia
4. Materials and Methods
4.1. Experimental Animals and Sample Collection
4.2. RNA Extraction, Transcriptome Sequencing, Quality Control, and Alignment
4.3. DEGs and Enrichment Analysis
4.4. Skin Tissue Nucleus Extraction and CUT&Tag Library Construction and Sequencing
4.5. CUT&Tag Sequencing Data Analysis
4.6. Chromatin State and Joint Transcriptome Analysis
4.7. Motif and Transcription Factor Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, J.; Fang, W.; Li, J.; Cai, W.; Lu, J. Full-length transcriptome reveals alternative splicing regulation pattern of skin color variant in red tilapia (Oreochromis spp.). Aquaculture 2025, 598, 741963. [Google Scholar] [CrossRef]
- Pradeep, P.J.; Srijaya, T.C.; Hassan, A.; Chatterji, A.K.; Withyachumnarnkul, B.; Jeffs, A. Optimal conditions for cold-shock induction of triploidy in red tilapia. Aquacult. Int. 2014, 22, 1163–1174. [Google Scholar] [CrossRef]
- Sugimoto, M. Morphological color changes in fish: Regulation of pigment cell density and morphology. Microsc. Res. Tech. 2002, 58, 496–503. [Google Scholar] [CrossRef]
- Fang, W.; Huang, J.; Li, S.; Lu, J. Identification of pigment genes (melanin, carotenoid and pteridine) associated with skin color variant in red tilapia using transcriptome analysis. Aquaculture 2022, 547, 737429. [Google Scholar] [CrossRef]
- Miyadai, M.; Takada, H.; Shiraishi, A.; Kimura, T.; Watakabe, I.; Kobayashi, H.; Nagao, Y.; Naruse, K.; Higashijima, S.; Shimizu, T.; et al. A gene regulatory network combining Pax3/7, Sox10 and Mitf generates diverse pigment cell types in medaka and zebrafish. Development 2023, 150, dev202114. [Google Scholar] [CrossRef]
- Leclercq, E.; Taylor, J.F.; Migaud, H. Morphological skin colour changes in teleosts. Fish Fish. 2010, 11, 159–193. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, L.; Dong, Z.; Chen, X.; Song, F.; Liu, N.; Yang, H.; Fu, J. Comparative Transcriptome Analysis Identifies Candidate Genes Related to Skin Color Differentiation in Red Tilapia. Sci. Rep. 2016, 6, 31347. [Google Scholar] [CrossRef]
- O’Quin, C.T. Mapping of pigmentation QTL on an anchored genome assembly of the cichlid fish, Metriaclima zebra. BMC Genom. 2013, 14, 287. [Google Scholar] [CrossRef]
- Berry, S.D.; Davis, S.R.; Beattie, E.M.; Thomas, N.L.; Burrett, A.K.; Ward, H.E.; Stanfield, A.M.; Biswas, M.; Ankersmit-Udy, A.E.; Oxley, P.E. Mutation in Bovine β-Carotene Oxygenase 2 Affects Milk Color. Genetics 2009, 182, 923–926. [Google Scholar] [CrossRef]
- Judan Cruz, K.G.; Landingin, E.P.; Gajeton, M.B.; Fernando, S.I.D.; Watanabe, K. Carotenoid coloration and coloration-linked gene expression in red tilapia (Oreochromis sp.) tissues. BMC Vet. Res. 2021, 17, 314. [Google Scholar] [CrossRef]
- Lanmei, W.; Wenbin, Z.; Zaijie, D.; Feibiao, S.; Juanjuan, D.; Jianjun, F. Comparative microRNA-seq Analysis Depicts Candidate miRNAs Involved in Skin Color Differentiation in Red Tilapia. Int. J. Mol. Sci. 2018, 19, 1209. [Google Scholar] [CrossRef]
- Wang, L.; Song, F.; Zhu, W.; Dong, J.; Fu, J.; Dong, Z. Effects of temperature on body color of Malaysian red tilapia during overwintering period. J. Fish. China 2018, 42, 72–79. [Google Scholar] [CrossRef]
- Wang, L.-m.; Luo, M.-k.; Yin, H.-r.; Zhu, W.-b.; Fu, J.-j.; Dong, Z.-j. Effects of background adaptation on the skin color of Malaysian red tilapia. Aquaculture 2020, 521, 735061. [Google Scholar] [CrossRef]
- Bhaumik, S.R.; Smith, E.; Shilatifard, A. Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007, 14, 1008–1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Fang, D. Overview of Histone Modification. In Histone Mutations and Cancer; Springer: Singapore, 2021; Volume 1283, pp. 1–16. [Google Scholar] [CrossRef]
- Wang, Z.; Ren, B. Role of H3K4 monomethylation in gene regulation. Curr. Opin. Genet. Dev. 2024, 84, 102153. [Google Scholar] [CrossRef]
- Rada-Iglesias, A. Is H3K4me1 at enhancers correlative or causative? Nat. Genet. 2018, 50, 4–5. [Google Scholar] [CrossRef]
- Kubo, N.; Chen, P.B.; Hu, R.; Ye, Z.; Sasaki, H.; Ren, B. H3K4me1 facilitates promoter-enhancer interactions and gene activation during embryonic stem cell differentiation. Mol. Cell 2024, 84, 17. [Google Scholar] [CrossRef]
- Sotomayor-Lugo, F.; Iglesias-Barrameda, N.; Castillo-Aleman, Y.M.; Casado-Hernandez, I.; Villegas-Valverde, C.A.; Bencomo-Hernandez, A.A.; Ventura-Carmenate, Y.; Rivero-Jimenez, R.A. The Dynamics of Histone Modifications during Mammalian Zygotic Genome Activation. Int. J. Mol. Sci. 2024, 25, 1459. [Google Scholar] [CrossRef]
- Wang, H.; Fan, Z.; Shliaha, P.V.; Miele, M.; Hendrickson, R.C.; Jiang, X.; Helin, K. Publisher Correction: H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 2023, 616, E7. [Google Scholar] [CrossRef]
- Beacon, T.H.; Delcuve, G.P.; López, C.; Nardocci, G.; Kovalchuk, I.; van Wijnen, A.J.; Davie, J.R. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin. Epigenet. 2021, 13, 138. [Google Scholar] [CrossRef]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H.; Roeder, R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef]
- Zhang, S.; Postnikov, Y.; Lobanov, A.; Furusawa, T.; Deng, T.; Bustin, M. H3K27ac nucleosomes facilitate HMGN localization at regulatory sites to modulate chromatin binding of transcription factors. Commun. Biol. 2022, 5, 159. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, X.; Wang, D.; Ma, J.; Wang, Y.; Wang, R.; Wang, H.; Cheng, W.; Zhu, Y. A narrative review of epigenetic marker in H3K27ac and its emerging potential as a therapeutic target in cancer. Epigenomics 2025, 17, 263–279. [Google Scholar] [CrossRef]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.-L.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef]
- Young, M.D.; Willson, T.A.; Wakefield, M.J.; Trounson, E.; Hilton, D.J.; Blewitt, M.E.; Oshlack, A.; Majewski, I.J. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011, 39, 7415–7427. [Google Scholar] [CrossRef]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Moose, H.E.; El-Hodiri, H.M.; Henion, P.D. colgate/hdac1 repression of foxd3 expression is required to permit mitfa-dependent melanogenesis. Dev. Biol. 2008, 313, 568–583. [Google Scholar] [CrossRef]
- Greenhill, E.R.; Rocco, A.; Vibert, L.; Nikaido, M.; Kelsh, R.N. An Iterative Genetic and Dynamical Modelling Approach Identifies Novel Features of the Gene Regulatory Network Underlying Melanocyte Development. PLoS Genet. 2011, 7, e1002265. [Google Scholar] [CrossRef]
- Raja, D.A.; Burse, V.; Subramaniam, S.A.; Sultan, Y.J.; Vats, F.; Gautam, A.; Sharma, H.; Sharma, B.; Singh, S.; Sivasubbu, A.; et al. pH-controlled histone acetylation amplifies melanocyte differentiation downstream of MITF. EMBO Rep. 2020, 21, e48333. [Google Scholar] [CrossRef]
- Tan, C.; Shi, C.; Li, Y.; Teng, W.; Li, Y.; Fu, H.; Ren, L.; Yu, H.; Li, Q.; Liu, S. Comparative Methylome Analysis Reveals Epigenetic Signatures Associated with Growth and Shell Color in the Pacific Oyster, Crassostrea gigas. Mar. Biotechnol. 2022, 24, 911–926. [Google Scholar] [CrossRef]
- Rzepka, Z.; Buszman, E.; Beberok, A.; Wrześniok, D. From tyrosine to melanin: Signaling pathways and factors regulating melanogenesis. Postpy Hig. I Med. Dowiadczalnej (Adv. Hyg. Exp. Med.) 2016, 70, 695–708. [Google Scholar] [CrossRef]
- Bejaoui, M.; Villareal, M.O.; Isoda, H. 3,4,5-Tri-O-Caffeoylquinic Acid Promoted Hair Pigmentation Through β-Catenin and Its Target Genes. Front. Cell Dev. Biol. 2020, 8, 175. [Google Scholar] [CrossRef]
- Guo, H.; Yang, K.; Deng, F.; Ye, J.; Xing, Y.; Li, Y.; Lian, X.; Yang, T. Wnt3a promotes melanin synthesis of mouse hair follicle melanocytes. Biochem. Biophys. Res. Commun. 2012, 420, 799–804. [Google Scholar] [CrossRef]
- Hsieh, C.-C.; Su, Y.-C.; Jiang, K.-Y.; Ito, T.; Li, T.-W.; Kaku-Ito, Y.; Cheng, S.-T.; Chen, L.-T.; Hwang, D.-Y.; Shen, C.-H. TRPM1 promotes tumor progression in acral melanoma by activating the Ca2+/CaMKIIδ/AKT pathway. J. Adv. Res. 2023, 43, 45–57. [Google Scholar] [CrossRef]
- Jiang, K.; Yu, H.; Kong, L.; Liu, S.; Li, Q. Molecular characterization of transcription factor CREB3L2 and CREB3L3 and their role in melanogenesis in Pacific oysters (Crassostrea gigas). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2024, 273, 110970. [Google Scholar] [CrossRef]
- Grichnik, J.M.; Burch, J.A.; Burchette, J.; Shea, C.R. The SCF/KIT pathway plays a critical role in the control of normal human melanocyte homeostasis. J. Investig. Dermatol. 1998, 111, 233–238. [Google Scholar] [CrossRef]
- Saldana-Caboverde, A.; Kos, L. Roles of endothelin signaling in melanocyte development and melanoma. Pigm. Cell Melanoma Res. 2010, 23, 160–170. [Google Scholar] [CrossRef]
- Szczygieł, M.; Müller-Decker, K.; Lucarelli, P.; Wäsch, M. Downregulation of the TGF-β pseudoreceptor BAMBI in non-small cell lung cancer enhances TGF-β signaling and invasion. Cancer Res. 2016, 76, 3785. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Si, L.; Tang, Q.; Lin, D.; Kong, X. Gain-of-Function Mutation of KIT Ligand on Melanin Synthesis Causes Familial Progressive Hyperpigmentation. Am. J. Hum. Genet. 2009, 84, 672–677. [Google Scholar] [CrossRef]
- Powell, M.B.; Hyman, P.; Bell, O.D.; Balmain, A.; Bowden, G.T. Hyperpigmentation and melanocytic hyperplasia in transgenic mice expressing the human T24 HA-ras gene regulated by a mouse tyrosinase promoter. Mol. Carcinog. 2010, 12, 82–90. [Google Scholar] [CrossRef]
- Moon, K.M.; Lee, B.; Kim, D.H.; Chung, H.Y. FoxO6 inhibits melanogenesis partly by elevating intracellular antioxidant capacity. Redox Biol. 2020, 36, 101624. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Passeron, T.; Watabe, H.; Yasumoto, K.I.; Rouzaud, F.; Hoashi, T.; Hearing, V.J. The effects of dickkopf 1 on gene expression and Wnt signaling by melanocytes: Mechanisms underlying its suppression of melanocyte function and proliferation. J. Investig. Dermatol. 2007, 127, 1217–1225. [Google Scholar] [CrossRef]
- Serre, C.; Busuttil, V.; Botto, J.a. Intrinsic and extrinsic regulation of human skin melanogenesis and pigmentation. Int. J. Cosmet. Sci. 2018, 40, 328–347. [Google Scholar] [CrossRef]
- Yang, G.; Li, Y.; Nishimura, E.K.; Xin, H.; Cui, R. Inhibition of PAX3 by TGF-β Modulates Melanocyte Viability. Mol. Cell 2008, 32, 554–563. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef]
- Klaassen, H.; Wang, Y.; Adamski, K.; Rohner, N.; Kowalko, J.E. CRISPR mutagenesis confirms the role of oca2 in melanin pigmentation in Astyanax mexicanus. Dev. Biol. 2018, 441, 313–318. [Google Scholar] [CrossRef]
- Javadiyan, S. Non-Synonymous variants in Premelanosome Protein (PMEL) cause ocular pigment dispersion and pigmentary glaucoma. Hum. Mol. Genet. 2019, 28, 1298–1311. [Google Scholar] [CrossRef]
- Park, P.J.; Lee, T.R.; Cho, E.G. Substance P Stimulates Endothelin 1 Secretion via Endothelin-Converting Enzyme 1 and Promotes Melanogenesis in Human Melanocytes. J. Investig. Dermatol. 2015, 135, 551–559. [Google Scholar] [CrossRef]
- Tanwar, J.; Ahuja, K.; Sharma, A.; Sehgal, P.; Ranjan, G.; Sultan, F.; Priya, A.; Venkatesan, M.; Yenamandra, V.K.; Singh, A. Mitochondrial calcium signaling mediated transcriptional regulation of keratin filaments is a critical determinant of melanogenesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Seberg, B.H.E.; Otterloo, E.V.; Loftus, S.K.; Liu, H.; Bonde, G.; Sompallae, R.; Gildea, D.E.; Santana, J.F.; Manak, J.R.; Pavan, W.J. TFAP2 paralogs regulate melanocyte differentiation in parallel with MITF. PLoS Genet. 2017, 13, e1006636. [Google Scholar] [CrossRef]
- Wang, Z.; Nishimura, Y.; Shimada, Y.; Umemoto, N.; Hirano, M.; Zang, L.; Oka, T.; Sakamoto, C.; Kuroyanagi, J.; Tanaka, T. Zebrafish β-adrenergic receptor mRNA expression and control of pigmentation. Gene 2009, 446, 18–27. [Google Scholar] [CrossRef]
- Thomas, A.J.; Erickson, C.A. FOXD3 regulates the lineage switch between neural crest-derived glial cells and pigment cells by repressing MITF through a non-canonical mechanism. Development 2009, 136, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Matsuda, M.; Yamashita, Y.; Takehara, M.; Fukuya, M.; Mineda, K.; Maji, D.; Ihn, H.; Adachi, H.; Sobue, G. Suppression of Melanin Production by Expression of HSP70. J. Biol. Chem. 2010, 285, 13254–13263. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Rao, C.; Li, H.; Chen, Y.; Hou, L. Regulation of pigmentation by microRNAs: MITF-dependent microRNA-211 targets TGF-β receptor 2. Pigm. Cell Melanoma Res. 2015, 28, 217–222. [Google Scholar] [CrossRef]
- Nakamasu, A.; Takahashi, G.; Kanbe, A.; Kondo, S. Interactions between zebrafish pigment cells responsible for the generation of Turing patterns. Proc. Natl. Acad. Sci. USA 2009, 106, 8429–8434. [Google Scholar] [CrossRef]
- Gaudel, C.; Soysouvanh, F.; Leclerc, J.; Bille, K.; Husser, C.; Montcriol, F.; Bertolotto, C.; Ballotti, R. Regulation of Melanogenesis by the Amino Acid Transporter SLC7A5–ScienceDirect. J. Investig. Dermatol. 2020, 140, 2253–2259. [Google Scholar] [CrossRef]
- Eda, A.; Enomoto, A.; Ikeda, M.; Ishiwatari, S.; Suzuki, T.; Matsukuma, S. Melanin pigmentation enhanced by the inhibition of Arginase-1 activity. J. Dermatol. Sci. 2016, 84, e86. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Lau, A.N.; Fiske, B.P.; Heiden, M.G.V. Increased PHGDH expression promotes aberrant melanin accumulation. BMC Cancer 2019, 19, 723. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Mesenchymal-epithelial interactions in the skin: Increased expression of dickkopf1 by palmoplantar fibroblasts inhibits melanocyte growth and differentiation. J. Cell Biol. 2004, 165, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Hasse, S.; Rokos, H.; Chavan, B.; Shalbaf, M.; Spencer, J.D.; Wood, J.M. Cholesterol regulates melanogenesis in human epidermal melanocytes and melanoma cells. Exp. Dermatol. 2009, 18, 680–688. [Google Scholar] [CrossRef]
- Dong, X.-R.; Wan, S.-M.; Zhou, J.-J.; Nie, C.-H.; Chen, Y.-L.; Diao, J.-H.; Gao, Z.-X. Functional differentiation of BMP7 genes in zebrafish: bmp7a for dorsal-ventral pattern and bmp7b for melanin synthesis and eye development. Front. Cell Dev. Biol. 2022, 10, 838721. [Google Scholar] [CrossRef] [PubMed]
- Bagati, A.; Bianchi-Smiraglia, A.; Moparthy, S.; Kolesnikova, K.; Nikiforov, M.A. FOXQ1 controls the induced differentiation of melanocytic cells. Cell Death Differ. 2018, 25, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, H.J.; Lee, A.Y. Aquaporin-3 Downregulation in Vitiligo Keratinocytes Increases Oxidative Stress of Melanocytes. Biomol. Ther. 2023, 31, 648–654. [Google Scholar] [CrossRef]
- Kim, J.Y.; Shin, J.Y.; Kim, M.; Hann, S.K.; Oh, S.H. Expression of cytosolic NADP+-dependent isocitrate dehydrogenase in melanocytes and its role as an antioxidant. J. Dermatol. Sci. 2012, 65, 118–125. [Google Scholar] [CrossRef]
- She, Q.; Dong, Y.; Li, D.; An, R.; Zhou, T.; Nie, X.; Pan, R.; Deng, Y. ABCB6 knockdown suppresses melanogenesis through the GSK3-β/β-catenin signaling axis in human melanoma and melanocyte cell lines. J. Dermatol. Sci. 2022, 106, 101–110. [Google Scholar] [CrossRef]
- Kato, M.; Takeda, K.; Kawamoto, Y.; Tsuzuki, T.; Dai, Y.; Nakayama, S.; Toriyama, K.; Tamada, Y.; Takahashi, M.; Nakashima, I. RET tyrosine kinase enhances hair growth in association with promotion of melanogenesis. Oncogene 2001, 20, 7536–7541. [Google Scholar] [CrossRef]
- Swoboda, A.; Soukup, R.; Eckel, O.; Kinslechner, K.; Moriggl, R. STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pathway. Oncogene 2021, 40, 1091–1105. [Google Scholar] [CrossRef]
- Kim, A.; Yang, Y.; Lee, M.S.; Yoo, Y.D.; Lee, H.G.; Lim, J.S. NDRG2 gene expression in B16F10 melanoma cells restrains melanogenesis via inhibition of Mitf expression. Pigm. Cell Melanoma Res. 2008, 21, 653–664. [Google Scholar] [CrossRef]
- Zhou, J.; An, X.; Dong, J.; Wang, Y.; Zhong, H.; Duan, L.; Ling, J.; Ping, F.; Shang, J. IL-17 induces cellular stress microenvironment of melanocytes to promote autophagic cell apoptosis in vitiligo. FASEB J. 2018, 32, 4899–4916. [Google Scholar] [CrossRef]
- Fossum, S.L.; Mutolo, M.J.; Tugores, A.; Ghosh, S.; Harris, A. Ets Homologous Factor (EHF) has Critical Roles in Epithelial Dysfunction in Airway Disease. J. Biol. Chem. 2017, 292, 775304. [Google Scholar] [CrossRef]
- Scimeca, M.; Montanaro, M.; Bonfiglio, R.; Anemona, L.; Agrò, E.F.; Asimakopoulos, A.D.; Bei, R.; Manzari, V.; Urbano, N.; Giacobbi, E.; et al. The ETS Homologous Factor (EHF) Represents a Useful Immunohistochemical Marker for Predicting Prostate Cancer Metastasis. Diagnostics 2022, 12, 800. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Huang, X.; Ruan, X.; Shang, X.; Zhang, M.; Liu, L.; Wang, P.; An, P.; Lin, Y.; Yang, J. Cpeb4-mediated Dclk2 promotes neuronal pyroptosis induced by chronic cerebral ischemia through phosphorylation of Ehf. J. Cereb. Blood Flow Metab. 2024, 44, 1655–1673. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.S. The Role of the Transcription Factor Ets1 in Melanocyte Development. FIU Electron. Theses Diss. 2014, 1451. [Google Scholar] [CrossRef]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef]
- Shin, S.Y.; Choi, J.H.; Jung, E.; Gil, H.N.; Lim, Y.; Lee, Y.H. The EGR1–STAT3 Transcription Factor Axis Regulates α-MSH–induced Tyrosinase Gene Transcription in Melanocytes. J. Investig. Dermatol. 2019, 139, 1616–1619. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, L.; You, W.; Xu, J.; Dai, J.; Hua, D.; Zhang, R.; Yao, F.; Zhou, S.; Huang, W.; et al. PRDM1/BLIMP1 induces cancer immune evasion by modulating the USP22-SPI1-PD-L1 axis in hepatocellular carcinoma cells. Nat. Commun. 2022, 13, 7677. [Google Scholar] [CrossRef]
- Olesnicky, E.; Hernandez-Lagunas, L.; Artinger, K.B. prdm1a Regulates sox10 and islet1 in the development of neural crest and Rohon-Beard sensory neurons. Genesis 2010, 48, 656–666. [Google Scholar] [CrossRef]
- Matsui, S.; Granitto, M.; Buckley, M.; Ludwig, K.; Koigi, S.; Shiley, J.; Zacharias, W.J.; Mayhew, C.N.; Lim, H.W.; Iwafuchi, M. Pioneer and PRDM transcription factors coordinate bivalent epigenetic states to safeguard cell fate. Mol. Cell 2024, 84, 476–489. [Google Scholar] [CrossRef]
- Hartman, M.L.; Talar, B.; Noman, M.Z.; Gajos-Michniewicz, A.; Chouaib, S.; Czyz, M. Gene Expression Profiling Identifies Microphthalmia-Associated Transcription Factor (MITF) and Dickkopf-1 (DKK1) as Regulators of Microenvironment-Driven Alterations in Melanoma Phenotype. PLoS ONE 2014, 9, e95157. [Google Scholar] [CrossRef] [PubMed]
- Huggins, I.J.; Bos, T.; Gaylord, O.; Jessen, C.; Willert, K. The WNT target SP5 negatively regulates WNT transcriptional programs in human pluripotent stem cells. Nat. Commun. 2017, 8, 1034. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Babraham Bioinformatics-FastQC A Quality Control tool for High Throughput Sequence Data. 2013. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 September 2024).
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Yang, L.; Smyth, G.K.; Wei, S. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Mu, H.; Chen, J.; Huang, W.; Huang, G.; Deng, M.; Hong, S.; Ai, P.; Gao, C.; Zhou, H. OmicShare tools: A zero-code interactive online platform for biological data analysis and visualization. iMeta 2024, 3, e228. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Kaya-Okur, H.S.; Ahmad, K. Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife Sci. 2020, 9, e63274. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Li, H. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Meers, M.P.; Tenenbaum, D.; Henikoff, S. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin 2019, 12, 42. [Google Scholar] [CrossRef]
- Fidel, R.; Ryan, D.P.; Björn, G.; Vivek, B.; Fabian, K.; Richter, A.S.; Steffen, H.; Friederike, D.; Thomas, M. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Robinson, J.T. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ripley, B.D. The R project in statistical computing. MSOR Connections. Newsl. Ltsn Maths Stats Or Netw. 2001, 1, 23–25. [Google Scholar]
- Bailey, T.L.; Mikael, B.; Buske, F.A.; Martin, F.; Grant, C.E.; Luca, C.; Jingyuan, R.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Black_Peaks | Red_Peaks |
|---|---|---|
| H3K4me1 | 9806 | 22,822 |
| H3K4me3 | 2185 | 2334 |
| H3K27ac | 41,748 | 79,260 |
| H3K27me3 | 34,812 | 51,589 |
| Sample | Diff_Peaks | Diff_Genes |
|---|---|---|
| H3K4me1 | 3542 | 2765 |
| H3K4me3 | 564 | 562 |
| H3K27ac | 1175 | 1032 |
| H3K27me3 | 2274 | 1952 |
| Sample | Gene Name | Gene Annotation | log2FC | Function | Reference |
|---|---|---|---|---|---|
| H3K4me1 | ctnnb1 | catenin (cadherin-associated protein), beta 1 | 1.12 | promote | Bejaoui M, et al. [34] |
| H3K4me1 | wnt3 | wingless-type MMTV integration site family, member 3 | 1.39 | promote | Guo H, et al. [35] |
| H3K4me1 | LOC112846512 | calcium/calmodulin-dependent protein kinase type II delta chain-like | 1.49 | promote | Hsieh C-C, et al. [36] |
| H3K4me1 | creb3l2 | cAMP responsive element binding protein 3-like 2 | 1.31 | promote | Jiang K, et al. [37] |
| H3K4me1 | kitlg | kit ligand | 3.22 | promote | Grichnik J M, et al. [38] |
| H3K4me1 | LOC102082978 | endothelin-3 | 1.85 | promote | Saldana C A, et al. [39] |
| H3K4me1 | bambi | BMP and activin membrane-bound inhibitor (Xenopus laevis) homolog | 1.92 | promote | Marwitz S, et al. [40] |
| H3K27ac | kitlg | kit ligand | 2.96 | promote | Grichnik J M, et al. [38] |
| H3K27ac | kit | KIT proto-oncogene, receptor tyrosine kinase | 1.29 | promote | Wang Z Q, et al. [41] |
| H3K4me1 | nf1 | neurofibromin 1 | −5.15 | inhibit | Powell M B, et al. [42] |
| H3K27ac | foxo6b | forkhead box O6 b | −2.55 | inhibit | Moon K M, et al. [43] |
| H3K27me3 | LOC100698110 | dickkopf-related protein 1 | 2.47 | inhibit | Yamaguchi Y, et al. [44] |
| H3K27ac | tgfbr2l | transforming growth factor beta receptor-like | −2.93 | inhibit | Serre C, et al. [45] |
| H3K27me3 | LOC100700558 | mothers against decapentaplegic homolog 4 | 1.27 | inhibit | Yang G, et al. [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Z.; Liu, J.; Shi, J.; Yin, M.; Liu, J.; Fan, J.; Bao, Z.; Ye, Z.; Hu, J. Integrated Transcriptomic and Epigenomic Analysis Reveals Mechanisms Underlying Melanotic Spot Formation in Red Tilapia (Oreochromis spp.). Int. J. Mol. Sci. 2025, 26, 4370. https://doi.org/10.3390/ijms26094370
Qi Z, Liu J, Shi J, Yin M, Liu J, Fan J, Bao Z, Ye Z, Hu J. Integrated Transcriptomic and Epigenomic Analysis Reveals Mechanisms Underlying Melanotic Spot Formation in Red Tilapia (Oreochromis spp.). International Journal of Molecular Sciences. 2025; 26(9):4370. https://doi.org/10.3390/ijms26094370
Chicago/Turabian StyleQi, Zhangru, Jiaxiang Liu, Jiale Shi, Miaomiao Yin, Jialong Liu, Jiaxuan Fan, Zhenmin Bao, Zhi Ye, and Jingjie Hu. 2025. "Integrated Transcriptomic and Epigenomic Analysis Reveals Mechanisms Underlying Melanotic Spot Formation in Red Tilapia (Oreochromis spp.)" International Journal of Molecular Sciences 26, no. 9: 4370. https://doi.org/10.3390/ijms26094370
APA StyleQi, Z., Liu, J., Shi, J., Yin, M., Liu, J., Fan, J., Bao, Z., Ye, Z., & Hu, J. (2025). Integrated Transcriptomic and Epigenomic Analysis Reveals Mechanisms Underlying Melanotic Spot Formation in Red Tilapia (Oreochromis spp.). International Journal of Molecular Sciences, 26(9), 4370. https://doi.org/10.3390/ijms26094370