
A Transfer Learning Framework for Predicting and Interpreting Drug Responses via Single-Cell RNA-Seq Data

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Data Analysis and Clustering Analysis
2.2. Performance Evaluation
2.3. Impact of Key Hyperparameters on Model Performance
2.4. Analysis of Modeling Strategies
2.5. Pathways Attribution
3. Discussion
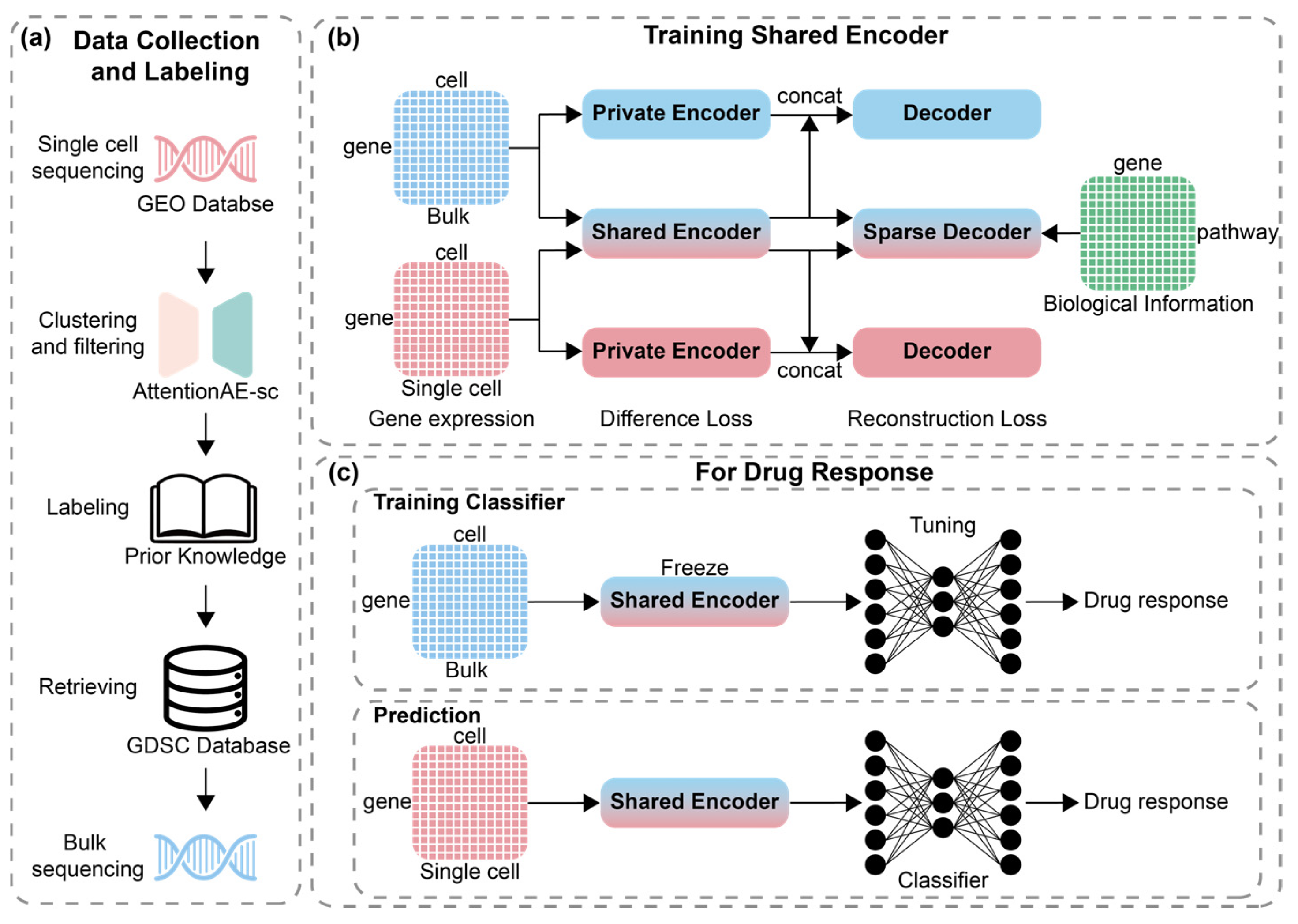
4. Materials and Methods
4.1. Data Collection and Processing
4.1.1. Collection
4.1.2. Labeling Strategy for Drug Response
4.2. Shared Encoder for Different Sequencing Data
4.3. Incorporating Biological Information with Sparse Decoder
4.4. Labels of Dataset and Training for Classifier
4.5. Model Performance Evaluation Metrics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| scRNA-seq | Single-cell RNA sequencing |
| DL | Deep learning |
| ML | Machine learning |
| RF | Random forest |
| VNN | Visible neural network |
| bulk RNA-seq | Bulk RNA sequencing |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| IG | Integrated gradients |
| IC50 | Half maximal inhibitory concentration |
| AUC | The area under the dose–response curve |
| ASW | Average silhouette width |
| UMAP | Uniform Manifold Approximation and Projection |
| LR | Logistic regression |
| SVM | Support vector machine |
| DT | Decision tree |
| RF | Random forest |
| GB | Gradient boosting |
| XGBoost | eXtreme Gradient Boosting |
| GO | Gene ontology |
| HVG | Highly variable gene |
| GEO | Gene Expression Omnibus |
References
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Li, L.; Peng, H.; Liu, L.; Heymann, D.; Robert, C.; Vallette, F.; Shen, S. Drug-Tolerant Persister Cells in Cancer: The Cutting Edges and Future Directions. Nat. Rev. Clin. Oncol. 2023, 20, 799–813. [Google Scholar] [CrossRef]
- Zheng, H.-C. The Molecular Mechanisms of Chemoresistance in Cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA Repair Pathways and Cisplatin Resistance: An Intimate Relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Bailey, C.; Shoura, M.J.; Mischel, P.S.; Swanton, C. Extrachromosomal DNA—Relieving Heredity Constraints, Accelerating Tumour Evolution. Ann. Oncol. 2020, 31, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in Biology and Targeted Therapy: New Insights and Translational Implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Yang, P.-L.; Liu, L.-X.; Li, E.-M.; Xu, L.-Y. STAT3, the Challenge for Chemotherapeutic and Radiotherapeutic Efficacy. Cancers 2020, 12, 2459. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Zhong, C.; Jiang, W.-J.; Yao, Y.; Li, Z.; Li, Y.; Wang, S.; Wang, X.; Zhu, W.; Wu, S.; Wang, J.; et al. CRISPR Screens Reveal Convergent Targeting Strategies against Evolutionarily Distinct Chemoresistance in Cancer. Nat. Commun. 2024, 15, 5502. [Google Scholar] [CrossRef]
- Gómez Tejeda Zañudo, J.; Barroso-Sousa, R.; Jain, E.; Jin, Q.; Li, T.; Buendia-Buendia, J.E.; Pereslete, A.; Abravanel, D.L.; Ferreira, A.R.; Wrabel, E.; et al. Exemestane plus Everolimus and Palbociclib in Metastatic Breast Cancer: Clinical Response and Genomic/Transcriptomic Determinants of Resistance in a Phase I/II Trial. Nat. Commun. 2024, 15, 2446. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Sedaghat-Hamedani, F.; Frese, K.; Hart, M.; Ludwig, N.; Meder, B.; Meese, E.; Keller, A. Bias in High-Throughput Analysis of miRNAs and Implications for Biomarker Studies. Anal. Chem. 2016, 88, 2088–2095. [Google Scholar] [CrossRef]
- Zhou, J.; Cipriani, A.; Liu, Y.; Fang, G.; Li, Q.; Cao, Y. Mapping Lesion-Specific Response and Progression Dynamics and Inter-Organ Variability in Metastatic Colorectal Cancer. Nat. Commun. 2023, 14, 417. [Google Scholar] [CrossRef]
- Gao, X.; Shen, W.; Ning, J.; Feng, Z.; Hu, J. Addressing Patient Heterogeneity in Disease Predictive Model Development. Biometrics 2022, 78, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [PubMed]
- Carli, F.; Di Chiaro, P.; Morelli, M.; Arora, C.; Bisceglia, L.; De Oliveira Rosa, N.; Cortesi, A.; Franceschi, S.; Lessi, F.; Di Stefano, A.L.; et al. Learning and Actioning General Principles of Cancer Cell Drug Sensitivity. Nat. Commun. 2025, 16, 1654. [Google Scholar] [CrossRef]
- Sharifi-Noghabi, H.; Zolotareva, O.; Collins, C.C.; Ester, M. MOLI: Multi-Omics Late Integration with Deep Neural Networks for Drug Response Prediction. Bioinformatics 2019, 35, i501–i509. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Chen, S.; Wang, J. DeepDRK: A Deep Learning Framework for Drug Repurposing through Kernel-Based Multi-Omics Integration. Brief. Bioinform. 2021, 22, bbab048. [Google Scholar] [CrossRef]
- He, D.; Liu, Q.; Wu, Y.; Xie, L. A Context-Aware Deconfounding Autoencoder for Robust Prediction of Personalized Clinical Drug Response from Cell-Line Compound Screening. Nat. Mach. Intell. 2022, 4, 879–892. [Google Scholar] [CrossRef]
- Kuenzi, B.M.; Park, J.; Fong, S.H.; Sanchez, K.S.; Lee, J.; Kreisberg, J.F.; Ma, J.; Ideker, T. Predicting Drug Response and Synergy Using a Deep Learning Model of Human Cancer Cells. Cancer Cell 2020, 38, 672–684.e6. [Google Scholar] [CrossRef]
- Huang, X.; Huang, K.; Johnson, T.; Radovich, M.; Zhang, J.; Ma, J.; Wang, Y. ParsVNN: Parsimony Visible Neural Networks for Uncovering Cancer-Specific and Drug-Sensitive Genes and Pathways. NAR Genom. Bioinform. 2021, 3, lqab097. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, H.; Li, S.; Leung, K.-S. Improving Prediction of Phenotypic Drug Response on Cancer Cell Lines Using Deep Convolutional Network. BMC Bioinform. 2019, 20, 408. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Z.; Jiang, R.; Zhou, M. DeepCDR: A Hybrid Graph Convolutional Network for Predicting Cancer Drug Response. Bioinform. Oxf. Engl. 2020, 36, i911–i918. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jiang, C.; Yu, X.; Fu, R.; Jin, S.; Liu, X. DeepTTA: A Transformer-Based Model for Predicting Cancer Drug Response. Brief. Bioinform. 2022, 23, bbac100. [Google Scholar] [CrossRef]
- Wu, Z.; Lawrence, P.J.; Ma, A.; Zhu, J.; Xu, D.; Ma, Q. Single-Cell Techniques and Deep Learning in Predicting Drug Response. Trends Pharmacol. Sci. 2020, 41, 1050–1065. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Shanahan, F.; Nguyen, T.T.T.; Staben, S.T.; Gazzard, L.; Yamazoe, S.; Wertz, I.E.; Piskol, R.; Yang, Y.A.; Modrusan, Z.; et al. Identifying Transcriptional Programs Underlying Cancer Drug Response with TraCe-Seq. Nat. Biotechnol. 2022, 40, 86–93. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- McFarland, J.M.; Paolella, B.R.; Warren, A.; Geiger-Schuller, K.; Shibue, T.; Rothberg, M.; Kuksenko, O.; Colgan, W.N.; Jones, A.; Chambers, E.; et al. Multiplexed Single-Cell Transcriptional Response Profiling to Define Cancer Vulnerabilities and Therapeutic Mechanism of Action. Nat. Commun. 2020, 11, 4296. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively Multiplex Chemical Transcriptomics at Single-Cell Resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef]
- Abdelaal, T.; Michielsen, L.; Cats, D.; Hoogduin, D.; Mei, H.; Reinders, M.J.T.; Mahfouz, A. A Comparison of Automatic Cell Identification Methods for Single-Cell RNA Sequencing Data. Genome Biol. 2019, 20, 194. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, H.; Zhang, S.; Li, Y.; Li, M. Attention-Based Deep Clustering Method for scRNA-Seq Cell Type Identification. PLOS Comput. Biol. 2023, 19, e1011641. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Sundararajan, M.; Taly, A.; Yan, Q. Axiomatic Attribution for Deep Networks. In Proceedings of the 34th International Conference on Machine Learning 2017, ICML’17, Sydney, NSW, Australia, 6–11 August 2017. [Google Scholar]
- Sayres, R.; Taly, A.; Rahimy, E.; Blumer, K.; Coz, D.; Hammel, N.; Krause, J.; Narayanaswamy, A.; Rastegar, Z.; Wu, D.; et al. Using a Deep Learning Algorithm and Integrated Gradients Explanation to Assist Grading for Diabetic Retinopathy. Ophthalmology 2019, 126, 552–564. [Google Scholar] [CrossRef]
- Qi, Z.; Khorram, S.; Fuxin, L. Visualizing Deep Networks by Optimizing with Integrated Gradients. Proc. AAAI Conf. Artif. Intell. 2020, 34, 11890–11898. [Google Scholar] [CrossRef]
- Zhuo, Y.; Ge, Z. IG2: Integrated Gradient on Iterative Gradient Path for Feature Attribution. IEEE Trans. Pattern Anal. Mach. Intell. 2024, 46, 7173–7190. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Jin, P.; Liu, J.; Li, S.; Liu, W.; Xi, S. Arsenic-Induced HER2 Promotes Proliferation, Migration and Angiogenesis of Bladder Epithelial Cells via Activation of Multiple Signaling Pathways in Vitro and in Vivo. Sci. Total Environ. 2021, 753, 141962. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Zhou, Z.; Jiang, C.; Bao, Z.; Wang, Y.; Zhang, Y.; Song, L.; Zhao, Y.; Li, X.; et al. The ATAC Complex Represses the Transcriptional Program of the Autophagy-Lysosome Pathway via Its E3 Ubiquitin Ligase Activity. Cell Rep. 2024, 43, 115033. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.e5. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhong, X.; Tan, Q.; Yang, S.; Liao, J.; Zhuge, J.; Hong, Z.; Deng, Q.; Zuo, Q. miR-301a-3p Induced by Endoplasmic Reticulum Stress Mediates the Occurrence and Transmission of Trastuzumab Resistance in HER2-Positive Gastric Cancer. Cell Death Dis. 2021, 12, 696. [Google Scholar] [CrossRef]
- Zhang, K.-R.; Zhang, Y.-F.; Lei, H.-M.; Tang, Y.-B.; Ma, C.-S.; Lv, Q.-M.; Wang, S.-Y.; Lu, L.-M.; Shen, Y.; Chen, H.-Z.; et al. Targeting AKR1B1 Inhibits Glutathione de Novo Synthesis to Overcome Acquired Resistance to EGFR-Targeted Therapy in Lung Cancer. Sci. Transl. Med. 2021, 13, eabg6428. [Google Scholar] [CrossRef]
- Figarol, S.; Delahaye, C.; Gence, R.; Doussine, A.; Cerapio, J.P.; Brachais, M.; Tardy, C.; Béry, N.; Asslan, R.; Colinge, J.; et al. Farnesyltransferase Inhibition Overcomes Oncogene-Addicted Non-Small Cell Lung Cancer Adaptive Resistance to Targeted Therapies. Nat. Commun. 2024, 15, 5345. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Ma, J.; Yu, M.K.; Fong, S.; Ono, K.; Sage, E.; Demchak, B.; Sharan, R.; Ideker, T. Using Deep Learning to Model the Hierarchical Structure and Function of a Cell. Nat. Methods 2018, 15, 290–298. [Google Scholar] [CrossRef]
- Seninge, L.; Anastopoulos, I.; Ding, H.; Stuart, J. VEGA Is an Interpretable Generative Model for Inferring Biological Network Activity in Single-Cell Transcriptomics. Nat. Commun. 2021, 12, 5684. [Google Scholar] [CrossRef]
- Tian, T.; Wan, J.; Song, Q.; Wei, Z. Clustering Single-Cell RNA-Seq Data with a Model-Based Deep Learning Approach. Nat. Mach. Intell. 2019, 1, 191–198. [Google Scholar] [CrossRef]
- Wang, J.; Ma, A.; Chang, Y.; Gong, J.; Jiang, Y.; Qi, R.; Wang, C.; Fu, H.; Ma, Q.; Xu, D. scGNN Is a Novel Graph Neural Network Framework for Single-Cell RNA-Seq Analyses. Nat. Commun. 2021, 12, 1882. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Drug | Number of Sensitive/ Resistant Cells | Number of Cells in GDSC |
|---|---|---|---|
| GSE117872 | Cisplatin | 950/352 | 735 |
| GSE131984 | Paclitaxel | 922/752 | 895 |
| GSE108394 | PLX-4720 | 3242/3236 | 898 |
| GSE156246_BT474 | Lapatinib | 714/1107 | 903 |
| GSE156246_HCC1419 | Lapatinib | 1584/4346 | 903 |
| Pathways Information Resource | Accuracy | F1 Score |
|---|---|---|
| Gene Ontology | 0.726 ± 0.008 | 0.841 ± 0.007 |
| Reactome | 0.710 ± 0.008 | 0.825 ± 0.007 |
| Hallmark | 0.696 ± 0.011 | 0.815 ± 0.009 |
| Methods | Accuracy | F1 Score |
|---|---|---|
| base AE | 0.2976 | 0.0834 |
| adv AE | 0.4545 | 0.3393 |
| base share AE | 0.3538 | 0.2407 |
| adv share AE | 0.5487 | 0.5145 |
| ours (share AE + pathways) | 0.7130 | 0.8308 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Li, S.; Lan, H.; Long, W.; Zhai, S.; Li, M.; Wen, Z. A Transfer Learning Framework for Predicting and Interpreting Drug Responses via Single-Cell RNA-Seq Data. Int. J. Mol. Sci. 2025, 26, 4365. https://doi.org/10.3390/ijms26094365
He Y, Li S, Lan H, Long W, Zhai S, Li M, Wen Z. A Transfer Learning Framework for Predicting and Interpreting Drug Responses via Single-Cell RNA-Seq Data. International Journal of Molecular Sciences. 2025; 26(9):4365. https://doi.org/10.3390/ijms26094365
Chicago/Turabian StyleHe, Yujie, Shenghao Li, Hao Lan, Wulin Long, Shengqiu Zhai, Menglong Li, and Zhining Wen. 2025. "A Transfer Learning Framework for Predicting and Interpreting Drug Responses via Single-Cell RNA-Seq Data" International Journal of Molecular Sciences 26, no. 9: 4365. https://doi.org/10.3390/ijms26094365
APA StyleHe, Y., Li, S., Lan, H., Long, W., Zhai, S., Li, M., & Wen, Z. (2025). A Transfer Learning Framework for Predicting and Interpreting Drug Responses via Single-Cell RNA-Seq Data. International Journal of Molecular Sciences, 26(9), 4365. https://doi.org/10.3390/ijms26094365