

Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression


Abstract
1. Overview of EMT
2. Types of EMT
2.1. EMT Type I
2.2. EMT Type II
2.3. EMT Type III
3. Transcription Factors That Induce EMT and Regulate Invasion and Metastasis
4. The Canonical Wnt-β-Catenin Signaling Pathway
5. The TGF-β Signaling Pathway
6. The Metastatic Cascade
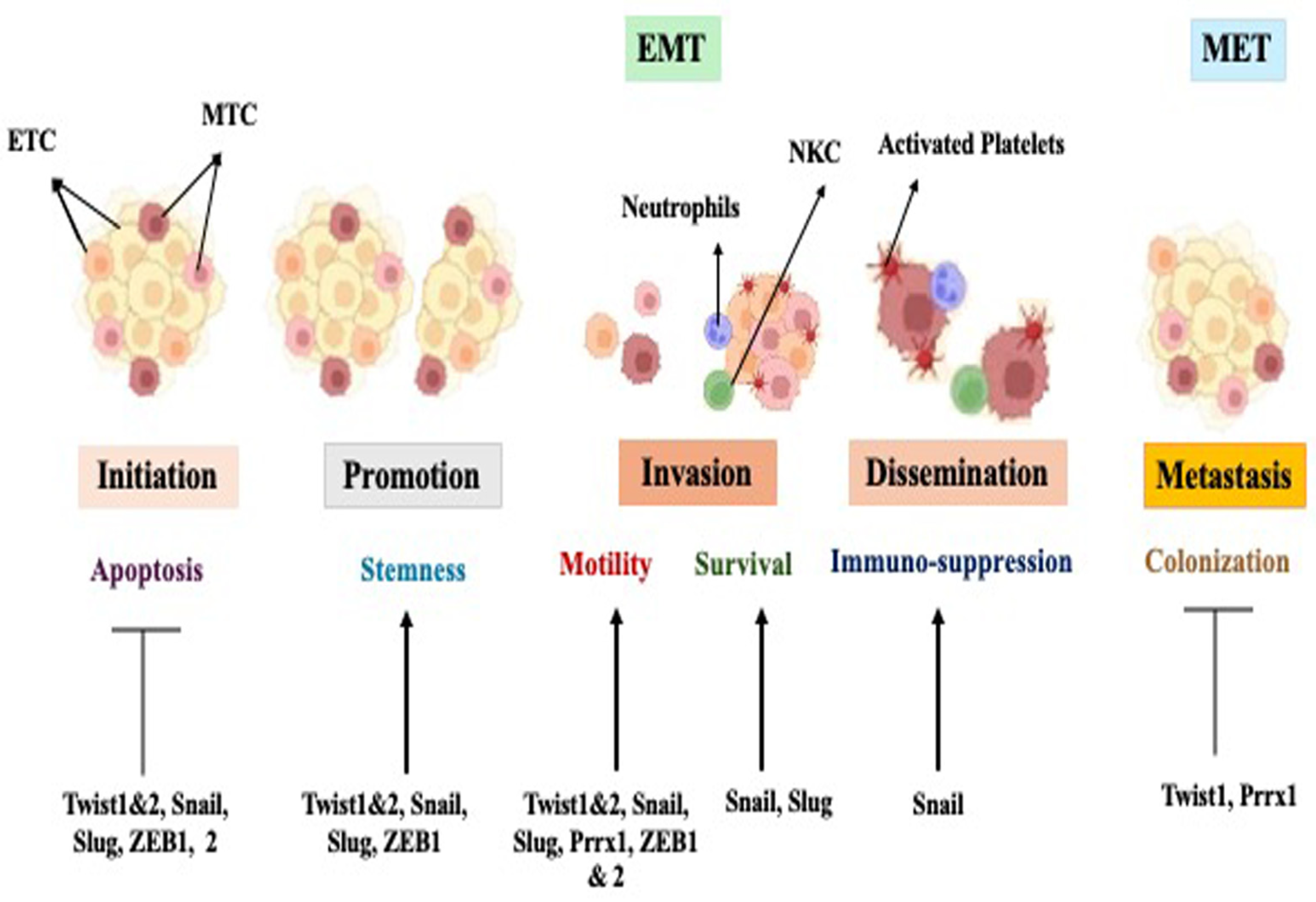
7. Role of EMT in Cancer Progression and Metastasis
Models of Metastasis
8. EMT and Circulating Tumor Cells (CTCs)
9. EMT and Tumor Angiogenesis
9.1. Impact of EMT and Angiogenesis on Metastasis
9.1.1. Metastatic Colonization
9.1.2. Resistance to Treatment
9.1.3. Endothelial–Mesenchymal Transition (EndMT)
10. EMT and Inflammation
11. Regulation and Modulation of EMT
11.1. Micro-Environmental Cues
11.1.1. Hypoxia as a Key EMT Regulator
11.1.2. Extracellular Matrix as a Regulator
11.2. Cellular Interactions
11.2.1. Cell–Cell Interactions
11.2.2. Paracrine Signaling
12. Therapeutic Strategies Targeting EMT
12.1. CRISPR/Cas9-Mediated Gene Editing
12.2. miRNA-Based Therapies
12.3. Exosome-Based Strategies
12.4. Immunotherapy
13. Conclusions
Funding
Conflicts of Interest
References
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, L. MicroRNA Control of Epithelial–Mesenchymal Transition and Metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef]
- Shaw, T.J.; Martin, P. Wound Repair: A Showcase for Cell Plasticity and Migration. Curr. Opin. Cell Biol. 2016, 42, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Pasupulati, A.K.; Nishad, R.; Nakuluri, K.; Motrapu, M. Epithelial–Mesenchymal Transition of Glomerular Podocytes: Implications in Proteinuria. MGM J. Med. Sci. 2017, 4, 26–34. [Google Scholar] [CrossRef]
- Deckert, T.; Feldt-Rasmussen, B.; Borch-Johnsen, K.; Jensen, T.; Kofoed-Enevoldsen, A. Albuminuria Reflects Widespread Vascular Damage. Diabetologia 1989, 32, 219–226. [Google Scholar] [CrossRef]
- Cao, Y. Lack of basic rationale in epithelial-mesenchymal transition and its related concepts. Cell Biosci. 2024, 14, 104. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-Mesenchymal Transitions: The Importance of Changing Cell State in Development and Disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Nawshad, A.; Hay, E.D. TGFβ3 Signaling Activates Transcription of the LEF1 Gene to Induce Epithelial Mesenchymal Transformation During Mouse Palate Development. J. Cell Biol. 2003, 163, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Kielbik, M.; Przygodzka, P.; Szulc-Kielbik, I.; Klink, M. Snail transcription factors as key regulators of chemoresistance, stemness and metastasis of ovarian cancer cells. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 189003. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt–Axin2–GSK3β Cascade Regulates Snail1 Activity in Breast Cancer Cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217, e20200388. [Google Scholar] [CrossRef]
- Tang, X.; Sui, X.; Weng, L.; Liu, Y. SNAIL1: Linking Tumor Metastasis to Immune Evasion. Front. Immunol. 2021, 12, 724200. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef]
- Sato, R.; Semba, T.; Saya, H.; Arima, Y. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells 2016, 34, 1997–2007. [Google Scholar] [CrossRef]
- Chuang, K.-T.; Chiou, S.-S.; Hsu, S.-H. Recent Advances in Transcription Factors Biomarkers and Targeted Therapies Focusing on Epithelial–Mesenchymal Transition. Cancers 2023, 15, 3338. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.-Y.; Tsai, M.-F.; Chang, T.-H.; Chang, Y.-L.; Yuan, A.; Yu, C.-J.; Lin, S.-B.; Liou, G.-Y.; Lee, M.-L.; Chen, J.J.; et al. Transcription Repressor Slug Promotes Carcinoma Invasion and Predicts Outcome of Patients with Lung Adenocarcinoma. Clin. Cancer Res. 2005, 11, 8070–8078. [Google Scholar] [CrossRef] [PubMed]
- Ru, G.-Q.; Wang, H.-J.; Xu, W.-J.; Zhao, Z.-S. Upregulation of Twist in Gastric Carcinoma Associated with Tumor Invasion and Poor Prognosis. Pathol. Oncol. Res. 2011, 17, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, N.; Xu, J.; Ding, Z.; Zong, R.; Liu, Z. Significance of Heterogeneous Twist2 Expression in Human Breast Cancers. PLoS ONE 2012, 7, e48178. [Google Scholar] [CrossRef]
- Si, W.; Huang, W.; Zheng, Y.; Yang, Y.; Liu, X.; Shan, L.; Zhou, X.; Wang, Y.; Su, D.; Gao, J.; et al. Dysfunction of the Reciprocal Feedback Loop between GATA3- and ZEB2-Nucleated Repression Programs Contributes to Breast Cancer Metastasis. Cancer Cell 2015, 27, 822–836. [Google Scholar] [CrossRef]
- Wang, G.; Dong, W.; Shen, H.; Mu, X.; Li, Z.; Lin, X.; Liu, Y.; Du, J. A Comparison of Twist and E-Cadherin Protein Expression in Primary Non-Small-Cell Lung Carcinoma and Corresponding Metastases. Eur. J. Cardio-Thoracic Surg. 2011, 39, 1028–1032. [Google Scholar] [CrossRef]
- Gasparotto, D.; Polesel, J.; Marzotto, A.; Colladel, R.; Piccinin, S.; Modena, P.; Grizzo, A.; Sulfaro, S.; Serraino, D.; Barzan, L.; et al. Overexpression of TWIST2 Correlates with Poor Prognosis in Head and Neck Squamous Cell Carcinomas. Oncotarget 2011, 2, 1165–1175. [Google Scholar] [CrossRef]
- Yang, M.-H.; Chang, S.-Y.; Chiou, S.-H.; Liu, C.-J.; Chi, C.-W.; Chen, P.-M.; Teng, S.-C.; Wu, K.-J. Overexpression of NBS1 Induces Epithelial–Mesenchymal Transition and Co-Expression of NBS1 and Snail Predicts Metastasis of Head and Neck Cancer. Oncogene 2007, 26, 1459–1467. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Tanaka, K.; Matsusita, K.; Fujikawa, H.; Saigusa, S.; Ohi, M.; Inoue, Y.; Mohri, Y.; Uchida, K.; et al. Clinical Significance of Zinc Finger E-Box Binding Homeobox 1 (ZEB1) in Human Gastric Cancer. J. Surg. Oncol. 2012, 106, 280–285. [Google Scholar] [CrossRef]
- Shin, N.R.; Jeong, E.H.; Choi, C.I.; Moon, H.J.; Kwon, C.H.; Chu, I.S.; Kim, G.H.; Jeon, T.Y.; Kim, D.H.; Lee, J.H.; et al. Overexpression of Snail Is Associated with Lymph Node Metastasis and Poor Prognosis in Patients with Gastric Cancer. BMC Cancer 2012, 12, 521. [Google Scholar] [CrossRef]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like Growth Factor-I–Dependent Up-Regulation of ZEB1 Drives Epithelial-to-Mesenchymal Transition in Human Prostate Cancer Cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Kwok, W.K.; Ling, M.-T.; Lee, T.-W.; Lau, T.C.; Zhou, C.; Zhang, X.; Chua, C.W.; Chan, K.W.; Chan, F.L.; Glackin, C.; et al. Up-Regulation of TWIST in Prostate Cancer and Its Implication as a Therapeutic Target. Cancer Res. 2005, 65, 5153–5162. [Google Scholar] [CrossRef]
- Elloul, S.; Silins, I.; Tropé, C.G.; Benshushan, A.; Davidson, B.; Reich, R. Expression of E-Cadherin Transcriptional Regulators in Ovarian Carcinoma. Virchows Arch. 2006, 449, 520–528. [Google Scholar] [CrossRef]
- Stacy, A.J.; Craig, M.P.; Sakaram, S.; Kadakia, M. ΔNp63α and microRNAs: Leveraging the epithelial-mesenchymal transition. Oncotarget 2017, 8, 2114–2129. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Su, Q.; Liu, H.; Wang, D.; Zhang, W.; Lu, Z.; Chen, Y.; Huang, X.; Li, W.; Zhang, C.; et al. Frizzled7 Promotes Epithelial-to-Mesenchymal Transition and Stemness Via Activating Canonical Wnt/β-Catenin Pathway in Gastric Cancer. Int. J. Biol. Sci. 2018, 14, 280–293. [Google Scholar] [CrossRef]
- Hinton, K.; Kirk, A.; Paul, P.; Persad, S. Regulation of the Epithelial to Mesenchymal Transition in Osteosarcoma. Biomolecules 2023, 13, 398. [Google Scholar] [CrossRef]
- Hüsemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Christofori, G. Epithelial–Mesenchymal Transition (EMT) and Metastasis: Yes, No, Maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef]
- Miyazono, K. Transforming Growth Factor-.BETA. Signaling in Epithelial-Mesenchymal Transition and Progression of Cancer. Proc. Jpn. Acad. Ser. B 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Gu, S.; Derynck, R.; Chen, Y.-G.; Feng, X.-H. New progress in roles of TGF-β signaling crosstalks in cellular functions, immunity and diseases. Cell Regen. 2024, 13, 11. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chen, R.; Lin, B.; Deng, R.; Liang, Y.; Zeng, J.; Ma, S.; Qiu, X. Cross-Talk between the TGF-β and Cell Adhesion Signaling Pathways in Cancer. Int. J. Med. Sci. 2024, 21, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Miranda, I.; Jahan, N.; Shevde, L.A. The metastatic cascade through the lens of therapeutic inhibition. Cell Rep. Med. 2025, 6, 101872. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel Progression of Primary Tumours and Metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Christofori, G. New Signals from the Invasive Front. Nature 2006, 441, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Kang, Y.; Massagué, J. Epithelial-Mesenchymal Transitions. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef]
- Wick, W.; Platten, M.; Weller, M. Glioma Cell Invasion: Regulation of Metalloproteinase Activity by TGF-Beta. J. Neuro-Oncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, J.; Yu, J.; Wu, Y.; Guo, J.; Xu, Z.; Sun, X. Knockdown of MMP-1 inhibits the progression of colorectal cancer by suppressing the PI3K/Akt/c-myc signaling pathway and EMT. Oncol. Rep. 2020, 43, 1103–1112. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Brabletz, T. To Differentiate or Not—Routes towards Metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the Hybrid Epithelial/Mesenchymal Phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Liu, B.; Situ, X.; Luo, Y.; Fu, T.; Wang, Y.; Xie, Z.; Ren, L.; Zhu, Y.; He, W.; et al. New insights into the correlations between circulating tumor cells and target organ metastasis. Signal Transduct. Target. Ther. 2023, 8, 465. [Google Scholar] [CrossRef] [PubMed]
- Loric, S.; Paradis, V.; Gala, J.-L.; Berteau, P.; Bedossa, P.; Benoit, G.; Eschwège, P. Abnormal E-Cadherin Expression and Prostate Cell Blood Dissemination as Markers of Biological Recurrence in Cancer. Eur. J. Cancer 2001, 37, 1475–1481. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic Signaling Pathways and Anti-Angiogenic Therapy for Cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Hu, Y.; He, M.-Y.; Zhu, L.-F.; Yang, C.-C.; Zhou, M.-L.; Wang, Q.; Zhang, W.; Zheng, Y.-Y.; Wang, D.-M.; Xu, Z.-Q.; et al. Tumor-Associated Macrophages Correlate with the Clinicopathological Features and Poor Outcomes via Inducing Epithelial to Mesenchymal Transition in Oral Squamous Cell Carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 12. [Google Scholar] [CrossRef]
- Fu, X.-T.; Dai, Z.; Song, K.; Zhang, Z.-J.; Zhou, Z.-J.; Zhou, S.-L.; Zhao, Y.-M.; Xiao, Y.-S.; Sun, Q.-M.; Ding, Z.-B.; et al. Macrophage-Secreted IL-8 Induces Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells by Activating the JAK2/STAT3/Snail Pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef]
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin. Cancer Biol. 2017, 47, 177–184. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.; Pillai, A.A.; Devi, A. Hypoxia Induced EMT: A Review on the Mechanism of Tumor Progression and Metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Hapke, R.Y.; Haake, S.M. Hypoxia-Induced Epithelial to Mesenchymal Transition in Cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A New Vision of Hypoxia Promoting Cancer Progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Kumar, S.; Das, A.; Sen, S. Extracellular Matrix Density Promotes EMT by Weakening Cell–Cell Adhesions. Mol. Biosyst. 2014, 10, 838–850. [Google Scholar] [CrossRef]
- Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The Extracellular Matrix in Tumor Progression and Metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular Matrix Remodeling in Tumor Progression and Immune Escape: From Mechanisms to Treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, K.; Szczylik, C.; Bielecka, Z.F.; Bartnik, E.; Porta, C.; Lian, F.; Czarnecka, A.M. The Role of the Cell–Cell Interactions in Cancer Progression. J. Cell. Mol. Med. 2015, 19, 283–296. [Google Scholar] [CrossRef]
- Pajic-Lijakovic, I.; Milivojevic, M.; McClintock, P.V.E. Physical aspects of epithelial cell–cell interactions: Hidden system complexities. Eur. Biophys. J. 2024, 53, 355–372. [Google Scholar] [CrossRef]
- Arias, A.M. Epithelial Mesenchymal Interactions in Cancer and Development. Cell 2001, 105, 425–431. [Google Scholar] [CrossRef]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M.; Haley, J.D. A Systems View of Epithelial–Mesenchymal Transition Signaling States. Clin. Exp. Metastasis 2011, 28, 137–155. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of Nitric Oxide-Mediated Inhibition of EMT in Cancer. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A Specific Inhibitor of TGF-β Receptor Kinase, SB-431542, as a Potent Antitumor Agent for Human Cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef]
- Arora, A.; Scholar, E.M. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef]
- Madhusudan, S.; Ganesan, T.S. Tyrosine Kinase Inhibitors in Cancer Therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Rosato, R.R.; Grant, S. Histone Deacetylase Inhibitors in Cancer Therapy. Cancer Biol. Ther. 2003, 2, 31–38. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Brueckner, B.; Kuck, D.; Lyko, F. DNA Methyltransferase Inhibitors for Cancer Therapy. Cancer J. 2007, 13, 17–22. [Google Scholar] [CrossRef]
- Casalino, L.; Verde, P. Multifaceted Roles of DNA Methylation in Neoplastic Transformation, from Tumor Suppressors to EMT and Metastasis. Genes 2020, 11, 922. [Google Scholar] [CrossRef] [PubMed]
- Mohammadinejad, R.; Biagioni, A.; Arunkumar, G.; Shapiro, R.; Chang, K.-C.; Sedeeq, M.; Taiyab, A.; Hashemabadi, M.; Pardakhty, A.; Mandegary, A.; et al. EMT Signaling: Potential Contribution of CRISPR/Cas Gene Editing. Cell. Mol. Life Sci. 2020, 77, 2701–2722. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Moradi-Chaleshtori, M.; Paryan, M.; Koochaki, A.; Sharifi, K.; Mohammadi-Yeganeh, S. Mesenchymal stem cell-derived exosomes enriched with miR-218 reduce the epithelial–mesenchymal transition and angiogenesis in triple-negative breast cancer cells. Eur. J. Med. Res. 2023, 28, 516. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Shin, E.; Seong, K.M.; Jin, Y.W.; Youn, H.; Youn, B. The Emerging Roles of Exosomes as EMT Regulators in Cancer. Cells 2020, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Increased Proteins | Decreased Proteins |
|---|---|
| N-cadherin | E-cadherin |
| Vimentin | Desmoplakin |
| N-cadherin | Occludin |
| MMP-9 | Cytokeratin |
| MMP-3 | |
| MMP-2 | |
| Fibronectin | |
| Snail 1 (Snail | |
| Snail 2 (Slug) | |
| Twist | |
| FOX C2 | |
| SOX 10 |
| Type of Cancer | Invasiveness/Metastasis | References |
|---|---|---|
| Lung | Slug, ZEB 1, Twist 1 | [20,22,23] |
| Breast | Snail, Slug, ZEB 1, ZEB 2, Twist 1, Twist 2 | [20,22,24,25,26] |
| Head and neck | Snail, Twist 2 | [27,28] |
| Gastric | Snail, ZEB 1, Twist 1 | [23,29,30] |
| Prostrate | ZEB 1, Twist 1 | [31,32] |
| Ovarian | ZEB 2 | [33] |
| Colorectal | Snail, Slug, ZEB 1, ZEB 2, Twist 1 | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafoor, S.; Garcia, E.; Jay, D.J.; Persad, S. Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression. Int. J. Mol. Sci. 2025, 26, 4364. https://doi.org/10.3390/ijms26094364
Ghafoor S, Garcia E, Jay DJ, Persad S. Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression. International Journal of Molecular Sciences. 2025; 26(9):4364. https://doi.org/10.3390/ijms26094364
Chicago/Turabian StyleGhafoor, Saima, Elizabeth Garcia, Daniel J. Jay, and Sujata Persad. 2025. "Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression" International Journal of Molecular Sciences 26, no. 9: 4364. https://doi.org/10.3390/ijms26094364
APA StyleGhafoor, S., Garcia, E., Jay, D. J., & Persad, S. (2025). Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression. International Journal of Molecular Sciences, 26(9), 4364. https://doi.org/10.3390/ijms26094364