
Glutamate-Based Therapeutic Strategies for Schizophrenia: Emerging Approaches Beyond Dopamine

Abstract
1. Introduction
2. Glutamate and Synaptic Plasticity
3. Glutamate and Cognitive Function
4. Glutamate Dysregulation in Schizophrenia
5. Neuron–Glia Interactions in Glutamate Dysregulation
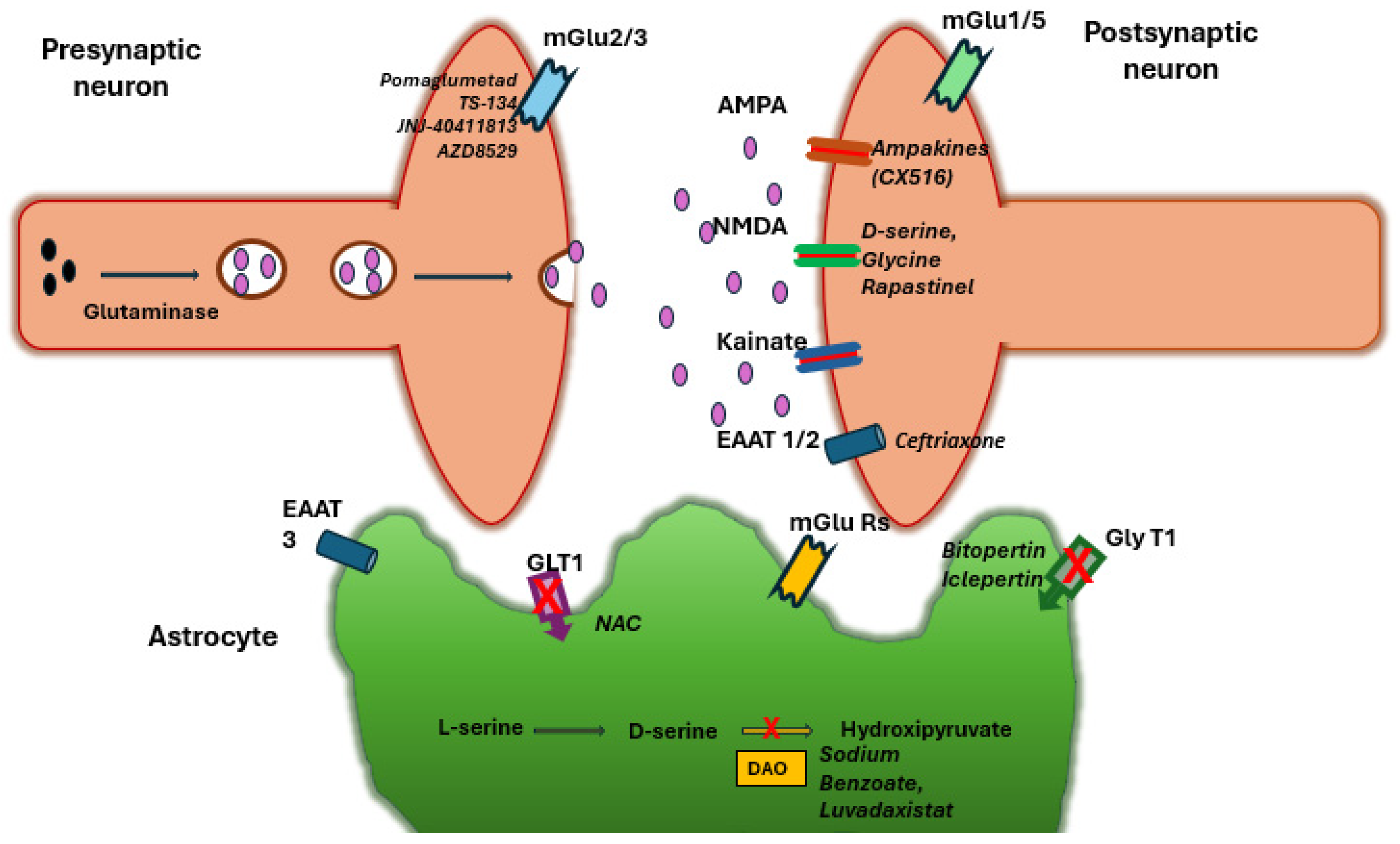
6. NMDA Receptor Modulators
6.1. Glycine and D-Serine (Co-Agonists)
6.2. Glycine Transporter-1 (GlyT1) Inhibitors
6.3. D-Amino Acid Oxidase (DAAO) Inhibitors
7. Metabotropic Glutamate Receptor Agents
7.1. Group II (mGlu2/3) Orthosteric Agonists
7.2. Group II Positive Allosteric Modulators (PAMs)
7.3. Group I (mGlu5) Modulators
7.4. Group III (mGlu4/8) Approaches
8. Glutamate Transporters and Homeostasis
8.1. EAAT2 Upregulation (Ceftriaxone and Others)
8.2. N-Acetylcysteine (NAC) and Redox Modulators
8.3. Other Glial and Synaptic Regulators
9. Kynurenine Pathway and Other Novel Strategies
9.1. Kynurenine Aminotransferase II (KAT II) Inhibitors
9.2. AMPA Receptor Modulators
9.2.1. Other Receptor Targets
9.2.2. Neuroplasticity and Others
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, V.; Moccia, F.; De Sarro, G.; Berra-Romani, R.; Soda, T.; Scarpellino, G.; Guerra, G. Two Signaling Modes Are Better than One: Flux-Independent Signaling by Ionotropic Glutamate Receptors Is Coming of Age. Biomedicines 2024, 12, 880. [Google Scholar] [CrossRef] [PubMed]
- Tarditi, A.M.; Klipfel, M.W.; Rodriguez, A.M.; Suvire, F.D.; Chassé, G.A.; Farkas, O.; Perczel, A.; Enriz, R.D. An Ab Initio Exploratory Study of Side Chain Conformations for Selected Backbone Conformations of N-Acetyl-L-glutamine-N-methylamide. J. Mol. Struct. (THEOCHEM) 2001, 545, 29–47. [Google Scholar] [CrossRef]
- Sedláček, M.; Vyklický, L.; Cais, O.; Kořínek, M.; Petrovič, M.; Chodounská, H.; Adamusová, E. Neurosteroid Modulation of Ionotropic Glutamate Receptors and Excitatory Synaptic Transmission. Physiol. Res. 2008, 57 (Suppl. S3), S49–S57. [Google Scholar] [CrossRef]
- Zhang, Y.; Chu, J.-M.-T.; Wong, G.-T.-C. Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules 2022, 12, 597. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Krystal, J.H.; Howes, O.D. Dopamine and Glutamate in Schizophrenia: Biology, Symptoms and Treatment. World Psychiatry 2020, 19, 15–33. [Google Scholar] [CrossRef]
- Demler, V.F.; Sterner, E.F.; Wilson, M.; Zimmer, C.; Knolle, F. The Impact of Spectral Basis Set Composition on Estimated Levels of Cingulate Glutamate and Its Associations with Different Personality Traits. BMC Psychiatry 2024, 24, 320. [Google Scholar] [CrossRef]
- Kennedy, M.B. Synaptic Signaling in Learning and Memory. Cold Spring Harb. Perspect. Biol. 2013, 8, a016824. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An Embarrassment of Riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Plaitakis, A.; Sidiropoulou, K.; Kotzamani, D.; Litso, I.; Zaganas, I.; Spanaki, C. Evolution of Glutamate Metabolism via GLUD2 Enhances Lactate-Dependent Synaptic Plasticity and Complex Cognition. Int. J. Mol. Sci. 2024, 25, 5297. [Google Scholar] [CrossRef]
- de León-López, C.A.M.; Carretero-Rey, M.; Khan, Z.U. AMPA Receptors in Synaptic Plasticity, Memory Function, and Brain Diseases. Cell. Mol. Neurobiol. 2025, 45, 14. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Peineau, S.; Howland, J.G.; Wang, Y.T. Long-Term Depression in the CNS. Nat. Rev. Neurosci. 2010, 11, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Song, M. Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target. Molecules 2024, 29, 5744. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic Alterations and Mitochondrial Impairment in a Murine Model of Alzheimer Disease. Neurobiol. Aging 2011, 33, 1121.e1–1121.e12. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Castillo, P.E. Heterosynaptic LTD of hippocampal GABAergic synapses: A novel role of endocannabinoids in regulating excitability. Neuron 2003, 38, 461–472. [Google Scholar] [CrossRef]
- Dong, B.; Yue, Y.; Dong, H.; Wang, Y. N-methyl-D-aspartate receptor hypofunction as a potential contributor to the progression and manifestation of many neurological disorders. Front. Mol. Neurosci. 2023, 16, 1174738. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, W.J.; Yang, S.S.; Mack, N.R.; Chamberlin, L.A. Aberrant Maturation and Connectivity of Prefrontal Cortex in Schizophrenia—Contribution of NMDA Receptor Development and Hypofunction. Mol. Psychiatry 2022, 27, 731–743. [Google Scholar] [CrossRef]
- Snyder, M.A.; Gao, W.J. NMDA Receptor Hypofunction for Schizophrenia Revisited: Perspectives from Epigenetic Mechanisms. Schizophr. Res. 2020, 217, 60–70. [Google Scholar] [CrossRef]
- Merritt, K.; Egerton, A.; Kempton, M.J.; Taylor, M.J.; McGuire, P.K. Nature of Glutamate Alterations in Schizophrenia: A Meta-Analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry 2016, 73, 665–674. [Google Scholar] [CrossRef]
- Egerton, A.; Grace, A.A.; Stone, J.; Bossong, M.G.; Sand, M.; McGuire, P. Glutamate in Schizophrenia: Neurodevelopmental Perspectives and Drug Development. Schizophr. Res. 2020, 223, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Kantrowitz, J.T. The Glutamate/N-Methyl-D-Aspartate Receptor (NMDAR) Model of Schizophrenia at 35: On the Path from Syndrome to Disease. Schizophr. Res. 2022, 242, 56–61. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Sandoval, C.; León-Ortiz, P.; Azcárraga, M.; Stephano, S.; Favila, R.; Díaz-Galvis, L.; Alvarado-Alanis, P.; Ramírez-Bermúdez, J.; Graff-Guerrero, A. Glutamate Levels in the Associative Striatum Before and After 4 Weeks of Antipsychotic Treatment in First-Episode Psychosis: A Longitudinal Proton Magnetic Resonance Spectroscopy Study. JAMA Psychiatry 2013, 70, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Bossong, M.G.; Antoniades, M.; Azis, M.; Samson, C.; Quinn, B.; Bonoldi, I.; Modinos, G.; Perez, J.; Howes, O.D.; Stone, J.M.; et al. Association of Hippocampal Glutamate Levels with Adverse Outcomes in Individuals at Clinical High Risk for Psychosis. JAMA Psychiatry 2019, 76, 199–207. [Google Scholar] [CrossRef]
- Bojesen, K.B.; Andersen, K.A.; Rasmussen, S.N.; Baandrup, L.; Madsen, L.M.; Glenthøj, B.Y.; Rostrup, E.; Broberg, B.V. Glutamate Levels and Resting Cerebral Blood Flow in Anterior Cingulate Cortex Are Associated at Rest and Immediately Following Infusion of S-Ketamine in Healthy Volunteers. Front. Psychiatry 2018, 9, 22. [Google Scholar] [CrossRef]
- Rowland, L.M.; Bustillo, J.R.; Mullins, P.G.; Jung, R.E.; Lenroot, R.; Landgraf, E.; Barrow, R.; Yeo, R.; Lauriello, J.; Brooks, W.M. Effects of Ketamine on Anterior Cingulate Glutamate Metabolism in Healthy Humans: A 4-T Proton MRS Study. Am. J. Psychiatry 2005, 162, 394–396. [Google Scholar] [CrossRef]
- Stone, J.M.; Dietrich, C.; Edden, R.; Mehta, M.A.; De Simoni, S.; Reed, L.J.; Krystal, J.H.; Nutt, D.; Barker, G.J. Ketamine Effects on Brain GABA and Glutamate Levels with 1H-MRS: Relationship to Ketamine-Induced Psychopathology. Mol. Psychiatry 2012, 17, 664–665. [Google Scholar] [CrossRef]
- Wenneberg, C.; Glenthøj, B.Y.; Hjorthøj, C.; Buchardt Zingenberg, F.J.; Glenthøj, L.B.; Rostrup, E.; Broberg, B.V.; Nordentoft, M. Cerebral Glutamate and GABA Levels in High-Risk of Psychosis States: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr. Res. 2020, 215, 38–48. [Google Scholar] [CrossRef]
- Egerton, A.; Stone, J.M.; Chaddock, C.A.; Barker, G.J.; Bonoldi, I.; Howard, R.M.; Merritt, K.; Allen, P.; Howes, O.D.; Murray, R.M.; et al. Relationship between Brain Glutamate Levels and Clinical Outcome in Individuals at Ultra High Risk of Psychosis. Neuropsychopharmacology 2014, 39, 2891–2899. [Google Scholar] [CrossRef]
- Demjaha, A.; Egerton, A.; Murray, R.M.; Kapur, S.; Howes, O.D.; Stone, J.M.; McGuire, P.K. Antipsychotic Treatment Resistance in Schizophrenia Associated with Elevated Glutamate Levels but Normal Dopamine Function. Biol. Psychiatry 2014, 75, e11–e13. [Google Scholar] [CrossRef]
- Egerton, A.; Brugger, S.; Raffin, M.; Barker, G.J.; Lythgoe, D.J.; McGuire, P.K.; Stone, J.M. Anterior Cingulate Glutamate Levels Related to Clinical Status Following Treatment in First-Episode Schizophrenia. Neuropsychopharmacology 2012, 37, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Nakajima, S.; Plitman, E.; Caravaggio, F.; Kim, J.; Shah, P.; Mar, W.; Chavez, S.; De Luca, V.; Mimura, M.; et al. Glutamatergic Neurometabolite Levels in Patients with Ultra-Treatment-Resistant Schizophrenia: A Cross-Sectional 3T Proton Magnetic Resonance Spectroscopy Study. Biol. Psychiatry 2019, 85, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Mouchlianitis, E.; Bloomfield, M.A.; Law, V.; Beck, K.; Selvaraj, S.; Rasquinha, N.; Waldman, A.; Turkheimer, F.E.; Egerton, A.; Stone, J.; et al. Treatment-Resistant Schizophrenia Patients Show Elevated Anterior Cingulate Cortex Glutamate Compared to Treatment-Responsive. Schizophr. Bull. 2016, 42, 744–752. [Google Scholar] [CrossRef]
- Tarumi, R.; Tsugawa, S.; Noda, Y.; Plitman, E.; Honda, S.; Matsushita, K.; Chavez, S.; Sawada, K.; Wada, M.; Matsui, M.; et al. Levels of glutamatergic neurometabolites in patients with severe treatment-resistant schizophrenia: A proton magnetic resonance spectroscopy study. Neuropsychopharmacology 2020, 45, 632–640. [Google Scholar] [CrossRef]
- Goldstein, M.E.; Anderson, V.M.; Pillai, A.; Kydd, R.R.; Russell, B.R. Glutamatergic Neurometabolites in Clozapine-Responsive and -Resistant Schizophrenia. Int. J. Neuropsychopharmacol. 2015, 18, pyu117. [Google Scholar] [CrossRef]
- Egerton, A.; Broberg, B.V.; Van Haren, N.; Merritt, K.; Barker, G.J.; Lythgoe, D.J.; Perez-Iglesias, R.; Baandrup, L.; Düring, S.W.; Sendt, K.V.; et al. Response to Initial Antipsychotic Treatment in First Episode Psychosis Is Related to Anterior Cingulate Glutamate Levels: A Multicentre 1H-MRS Study (OPTiMiSE). Mol. Psychiatry 2018, 23, 2145–2155. [Google Scholar] [CrossRef]
- Merritt, K.; Perez-Iglesias, R.; Sendt, K.V.; Goozee, R.; Jauhar, S.; Pepper, F.; Barker, G.J.; Glenthøj, B.; Arango, C.; Lewis, S.; et al. Remission from Antipsychotic Treatment in First Episode Psychosis Related to Longitudinal Changes in Brain Glutamate. NPJ Schizophr. 2019, 5, 12. [Google Scholar] [CrossRef]
- Jelen, L.A.; King, S.; Horne, C.M.; Lythgoe, D.J.; Young, A.H.; Stone, J.M. Functional Magnetic Resonance Spectroscopy in Patients with Schizophrenia and Bipolar Affective Disorder: Glutamate Dynamics in the Anterior Cingulate Cortex during a Working Memory Task. Eur. Neuropsychopharmacol. 2019, 29, 222–234. [Google Scholar] [CrossRef]
- Taylor, R.; Schaefer, B.; Densmore, M.; Neufeld, R.W.; Rajakumar, N.; Williamson, P.C.; Théberge, J. Increased Glutamate Levels Observed upon Functional Activation in the Anterior Cingulate Cortex Using the Stroop Task and Functional Spectroscopy. Neuroreport 2015, 26, 107–112. [Google Scholar] [CrossRef]
- Mei, Y.Y.; Wu, D.C.; Zhou, N. Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 2018, 9, 544. [Google Scholar] [CrossRef]
- Hashimoto, K.; Engberg, G.; Shimizu, E.; Nordin, C.; Lindström, L.H.; Iyo, M. Elevated Glutamine/Glutamate Ratio in Cerebrospinal Fluid of First Episode and Drug-Naive Schizophrenic Patients. BMC Psychiatry 2005, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.K.; Wu, T.H.; Chen, C.H.; Lu, M.L. Efficacy of N-Methyl-D-Aspartate Receptor Modulator Augmentation in Schizophrenia: A Meta-Analysis of Randomised, Placebo-Controlled Trials. J. Psychopharmacol. 2021, 35, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Malhotra, A.K.; Cornblatt, B.; Silipo, G.; Balla, A.; Suckow, R.F.; D’Souza, C.; Saksa, J.; Woods, S.W.; Javitt, D.C. High Dose D-Serine in the Treatment of Schizophrenia. Schizophr. Res. 2010, 121, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Rosenbrock, H.; Dorner-Ciossek, C.; Giovannini, R.; Schmid, B.; Schuelert, N. Effects of the Glycine Transporter-1 Inhibitor Iclepertin (BI 425809) on Sensory Processing, Neural Network Function, and Cognition in Animal Models Related to Schizophrenia. J. Pharmacol. Exp. Ther. 2022, 382, 223–232. [Google Scholar] [CrossRef]
- Moreno, J.L.; Miranda-Azpiazu, P.; García-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric Signaling through an mGlu2 and 5-HT2A Heteromeric Receptor Complex and Its Potential Contribution to Schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef]
- Stauffer, V.L.; Millen, B.A.; Andersen, S.; Kinon, B.J.; Lagrandeur, L.; Lindenmayer, J.P.; Gomez, J.C. Pomaglumetad Methionil: No Significant Difference as an Adjunctive Treatment for Patients with Prominent Negative Symptoms of Schizophrenia Compared to Placebo. Schizophr. Res. 2013, 150, 434–441. [Google Scholar] [CrossRef]
- Biso, L.; Carli, M.; Scarselli, M.; Longoni, B. Overview of Novel Antipsychotic Drugs: State of the Art, New Mechanisms, and Clinical Aspects of Promising Compounds. Biomedicines 2025, 13, 85. [Google Scholar] [CrossRef]
- Stennett, B.A.; Frankowski, J.C.; Peris, J.; Knackstedt, L.A. Ceftriaxone Reduces Alcohol Intake in Outbred Rats While Upregulating xCT in the Nucleus Accumbens Core. Pharmacol. Biochem. Behav. 2017, 159, 18–23. [Google Scholar] [CrossRef]
- Bai, M.Y.; Lovejoy, D.B.; Guillemin, G.J.; Kozak, R.; Stone, T.W.; Koola, M.M. Galantamine–Memantine Combination and Kynurenine Pathway Enzyme Inhibitors in the Treatment of Neuropsychiatric Disorders. Complex Psychiatry 2021, 7, 19–33. [Google Scholar] [CrossRef]
- Kaul, I.; Sawchak, S.; Correll, C.U.; Kakar, R.; Breier, A.; Zhu, H.; Miller, A.C.; Paul, S.M.; Brannan, S.K. Efficacy and Safety of the Muscarinic Receptor Agonist KarXT (Xanomeline–Trospium) in Schizophrenia (EMERGENT-2) in the USA: Results from a Randomised, Double-Blind, Placebo-Controlled, Flexible-Dose Phase 3 Trial. Lancet 2024, 403, 160–170. [Google Scholar] [CrossRef]
- Husain, M.O.; Chaudhry, I.B.; Khoso, A.B.; Husain, M.I.; Ansari, M.A.; Mehmood, N.; Naqvi, H.A.; Nizami, A.T.; Talib, U.; Rajput, A.H.; et al. Add-On Sodium Benzoate and N-Acetylcysteine in Patients with Early Schizophrenia Spectrum Disorder: A Multicenter, Double-Blind, Randomized Placebo-Controlled Feasibility Trial. Schizophr. Bull. Open 2024, 5, sgae004. [Google Scholar] [CrossRef] [PubMed]
- Réthelyi, J.M.; Vincze, K.; Schall, D.; Glennon, J.; Berkel, S. The Role of Insulin/IGF1 Signalling in Neurodevelopmental and Neuropsychiatric Disorders—Evidence from Human Neuronal Cell Models. Neurosci. Biobehav. Rev. 2023, 153, 105330. [Google Scholar] [CrossRef] [PubMed]
- Staats Pires, A.; Krishnamurthy, S.; Sharma, S.; Chow, S.; Klistorner, S.; Guillemin, G.J.; Klistorner, A.; You, Y.; Heng, B. Dysregulation of the Kynurenine Pathway in Relapsing Remitting Multiple Sclerosis and Its Correlations with Progressive Neurodegeneration. Neurol. Neuroimmunol. Neuroinflamm. 2025, 12, e200372. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Sapkota, K. The Origin of NMDA Receptor Hypofunction in Schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. Methods Mol. Biol. 2017, 1677, 1–80. [Google Scholar] [CrossRef]
- Chen, S.; Xu, D.; Fan, L.; Fang, Z.; Wang, X.; Li, M. Roles of N-Methyl-D-Aspartate Receptors (NMDARs) in Epilepsy. Front. Mol. Neurosci. 2022, 14, 797253. [Google Scholar] [CrossRef]
- Pei, J.-C.; Luo, D.-Z.; Gau, S.-S.; Chang, C.-Y.; Lai, W.-S. Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients with Schizophrenia. Front. Psychiatry 2021, 12, 742058. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, J.; Wu, R. Drugs Based on NMDAR Hypofunction Hypothesis in Schizophrenia. Front. Neurosci. 2021, 15, 641047. [Google Scholar] [CrossRef]
- Meftah, A.; Hasegawa, H.; Kantrowitz, J.T. D-Serine: A Cross-Species Review of Safety. Front. Psychiatry 2021, 12, 726365. [Google Scholar] [CrossRef]
- Guercio, G.D.; Panizzutti, R. Potential and Challenges for the Clinical Use of D-Serine as a Cognitive Enhancer. Front. Psychiatry 2018, 9, 14. [Google Scholar] [CrossRef]
- Goff, D.C. D-Cycloserine in Schizophrenia: New Strategies for Improving Clinical Outcomes by Enhancing Plasticity. Curr. Neuropharmacol. 2017, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Sur, C.; Kinney, G.G. Glycine Transporter 1 Inhibitors and Modulation of NMDA Receptor-Mediated Excitatory Neurotransmission. Curr. Drug Targets 2007, 8, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Umbricht, D.; Alberati, D.; Martin-Facklam, M.; Borroni, E.; Youssef, E.A.; Ostland, M.; Wallace, T.L.; Knoflach, F.; Dorflinger, E.; Wettstein, J.G.; et al. Effect of Bitopertin, a Glycine Reuptake Inhibitor, on Negative Symptoms of Schizophrenia: A Randomized, Double-Blind, Proof-of-Concept Study. JAMA Psychiatry 2014, 71, 637–646. [Google Scholar] [CrossRef]
- Bugarski-Kirola, D.; Blaettler, T.; Arango, C.; Fleischhacker, W.W.; Garibaldi, G.; Wang, A.; Dixon, M.; Bressan, R.A.; Nasrallah, H.; Lawrie, S.; et al. Bitopertin in Negative Symptoms of Schizophrenia—Results from the Phase III FlashLyte and DayLyte Studies. Biol. Psychiatry 2017, 82, 8–16. [Google Scholar] [CrossRef]
- Harvey, P.D.; McDonald, S.; Fu, E.; Reuteman-Fowler, C. Efficacy and Safety of Iclepertin (BI 425809) with Adjunctive Computerized Cognitive Training in Patients with Schizophrenia. Schizophr. Res. Cogn. 2024, 40, 100340. [Google Scholar] [CrossRef]
- Reuteman-Fowler, C.; Blahova, Z.; Ikezawa, S.; Marder, S.; Falkai, P.; Krystal, J.H. The Phase III CONNEX Programme Assessing the Efficacy and Safety of Iclepertin in Patients with Schizophrenia: Trial Design and Recruitment Update. Eur. Psychiatry 2024, 67 (Suppl. S1), S87–S88. [Google Scholar] [CrossRef]
- Boehringer Ingelheim. Top-Line Results from the Phase III CONNEX Clinical Program in Cognitive Impairment in Adults with Schizophrenia Show Primary and Key Secondary Endpoints Were Not Met. Medthority. 19 January 2025. Available online: https://www.medthority.com/news/2025/1/top-line-results-from-the-phase-iii-connex-clinical-program-in-cognitive-impairment-in-adults-with-schizophrenia-show-primary-and-key-secondary-endpoints-were-not-met---boehringer-ingelheim/ (accessed on 28 February 2025).
- Zhang, H.X.; Lyons-Warren, A.; Thio, L.L. The Glycine Transport Inhibitor Sarcosine Is an Inhibitory Glycine Receptor Agonist. Neuropharmacology 2009, 57, 551–555. [Google Scholar] [CrossRef]
- Nagy, L.V.; Bali, Z.K.; Kapus, G.; Pelsőczi, P.; Farkas, B.; Lendvai, B.; Lévay, G.; Hernádi, I. Converging Evidence on D-Amino Acid Oxidase-Dependent Enhancement of Hippocampal Firing Activity and Passive Avoidance Learning in Rats. Int. J. Neuropsychopharmacol. 2021, 24, 434–445. [Google Scholar] [CrossRef]
- Lane, H.Y.; Lin, C.H.; Green, M.F.; Hellemann, G.; Huang, C.C.; Chen, P.W.; Tun, R.; Chang, Y.C.; Tsai, G.E. Add-On Treatment of Benzoate for Schizophrenia: A Randomized, Double-Blind, Placebo-Controlled Trial of D-Amino Acid Oxidase Inhibitor. JAMA Psychiatry 2013, 70, 1267–1275. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, C.H.; Chang, Y.C.; Huang, Y.J.; Chen, P.W.; Yang, H.T.; Lane, H.Y. Sodium Benzoate, a D-Amino Acid Oxidase Inhibitor, Added to Clozapine for the Treatment of Schizophrenia: A Randomized, Double-Blind, Placebo-Controlled Trial. Biol. Psychiatry 2018, 84, 422–432. [Google Scholar] [CrossRef]
- Fradley, R.; Goetghebeur, P.; Miller, D.; Burley, R.; Almond, S.; Gruart, I.M.; Delgado García, J.M.; Zhu, B.; Howley, E.; Neill, J.C.; et al. Luvadaxistat: A novel potent and selective d-amino acid oxidase inhibitor improves cognitive and social deficits in rodent models of schizophrenia. Neurochem. Res. 2023, 48, 3027–3041. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.; Dong, C.; Murthy, V.; Asgharnejad, M.; Du, X.; Summerfelt, A.; Lu, H.; Xu, L.; Wendland, J.R.; Dunayevich, E.; et al. The D-Amino Acid Oxidase Inhibitor Luvadaxistat Improves Mismatch Negativity in Patients with Schizophrenia in a Randomized Trial. Neuropsychopharmacology 2023, 48, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.; Hanson, E.; DeMartinis, N.; Asgharnejad, M.; Dong, C.; Evans, R.; Ge, T.; Dunayevich, E.; Singh, J.B.; Ratti, E.; et al. INTERACT: A Randomized Phase 2 Study of the DAAO Inhibitor Luvadaxistat in Adults with Schizophrenia. Schizophr. Res. 2024, 270, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, C.; Zhong, N.; Wang, Y.; Huang, Y.; Zhang, X. Advances in the Treatment of Cognitive Impairment in Schizophrenia: Targeting NMDA Receptor Pathways. Int. J. Mol. Sci. 2024, 25, 10668. [Google Scholar] [CrossRef]
- Kato, T.; Duman, R. Rapastinel, a Novel Glutamatergic Agent with Ketamine-Like Antidepressant Actions: Convergent Mechanisms. Pharmacol. Biochem. Behav. 2019, 188, 172827. [Google Scholar] [CrossRef]
- Correll, C.U.; Solmi, M.; Cortese, S.; Fava, M.; Højlund, M.; Kraemer, H.C.; McIntyre, R.S.; Pine, D.S.; Schneider, L.S.; Kane, J.M. The Future of Psychopharmacology: A Critical Appraisal of Ongoing Phase 2/3 Trials, and of Some Current Trends Aiming to De-Risk Trial Programmes of Novel Agents. World Psychiatry 2023, 22, 48–74. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef]
- Kruse, A.O.; Bustillo, J.R. Glutamatergic Dysfunction in Schizophrenia. Transl. Psychiatry 2022, 12, 500. [Google Scholar] [CrossRef]
- Woodhall, G.; Evans, D.I.; Jones, R.S. Activation of Presynaptic Group III Metabotropic Glutamate Receptors Depresses Spontaneous Inhibition in Layer V of the Rat Entorhinal Cortex. Neuroscience 2001, 105, 71–78. [Google Scholar] [CrossRef]
- Moreno, J.L.; González-Maeso, J. Preclinical Models of Antipsychotic Drug Action. Int. J. Neuropsychopharmacol. 2013, 16, 2131–2144. [Google Scholar] [CrossRef]
- Adams, D.H.; Kinon, B.J.; Baygani, S.; Millen, B.A.; Velona, I.; Kollack-Walker, S.; Walling, D.P. A Long-Term, Phase 2, Multicenter, Randomized, Open-Label, Comparative Safety Study of Pomaglumetad Methionil (LY2140023 Monohydrate) versus Atypical Antipsychotic Standard of Care in Patients with Schizophrenia. BMC Psychiatry 2013, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Grinband, J.; Goff, D.C.; Lahti, A.; Marder, S.R.; Kegeles, L.S.; Girgis, R.R.; Sobeih, T.; Wall, M.M.; Choo, T.-H.; et al. Proof mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: RCTs of pomaglumetad and TS-134 on ketamine-induced psychotic symptoms and pharmacokinetics in healthy volunteers. Neuropsychopharmacology 2020, 45, 11. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Marcy, B.; Kinoshita, K.; Fukasawa, M.; Hikichi, H.; Chaki, S.; Okuyama, S.; Gevorkyan, H.; Yoshida, S. Safety and Pharmacokinetic Profiles of MGS0274 Besylate (TS-134), a Novel Metabotropic Glutamate 2/3 Receptor Agonist Prodrug, in Healthy Subjects. Br. J. Clin. Pharmacol. 2020, 86, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef]
- Lavreysen, H.; Ahnaou, A.; Drinkenburg, W.; Langlois, X.; Mackie, C.; Pype, S.; Lütjens, R.; Le Poul, E.; Trabanco, A.A.; Nuñez, J.M. Pharmacological and Pharmacokinetic Properties of JNJ-40411813, a Positive Allosteric Modulator of the mGlu2 Receptor. Pharmacol. Res. Perspect. 2015, 3, e00096. [Google Scholar] [CrossRef]
- Lavreysen, H.; Langlois, X.; Donck, L.V.; Nuñez, J.M.; Pype, S.; Lütjens, R.; Megens, A. Preclinical Evaluation of the Antipsychotic Potential of the mGlu2-Positive Allosteric Modulator JNJ-40411813. Pharmacol. Res. Perspect. 2015, 3, e00097. [Google Scholar] [CrossRef]
- Kang, W.; Frouni, I.; Kwan, C.; Bédard, D.; Nuara, S.G.; Hamadjida, A.; Gourdon, J.C.; Huot, P. Effect of the mGlu2 Positive Allosteric Modulator Biphenyl-Indanone A as a Monotherapy and as Adjunct to a Low Dose of L-DOPA in the MPTP-Lesioned Marmoset. Eur. J. Neurosci. 2024, 60, 6175–6184. [Google Scholar] [CrossRef]
- Litman, R.E.; Smith, M.A.; Doherty, J.J.; Cross, A.; Raines, S.; Gertsik, L.; Zukin, S.R. AZD8529, a Positive Allosteric Modulator at the mGluR2 Receptor, Does Not Improve Symptoms in Schizophrenia: A Proof of Principle Study. Schizophr. Res. 2016, 172, 152–157. [Google Scholar] [CrossRef]
- Wolf, D.H.; Zheng, D.; Kohler, C.; Turetsky, B.I.; Ruparel, K.; Satterthwaite, T.D.; Elliott, M.A.; March, M.E.; Cross, A.J.; Smith, M.A.; et al. Effect of mGluR2 Positive Allosteric Modulation on Frontostriatal Working Memory Activation in Schizophrenia. Mol. Psychiatry 2022, 27, 1226–1232. [Google Scholar] [CrossRef]
- Stansley, B.J.; Conn, P.J. The Therapeutic Potential of Metabotropic Glutamate Receptor Modulation for Schizophrenia. Curr. Opin. Pharmacol. 2018, 38, 31–36. [Google Scholar] [CrossRef]
- Doriat, J.F.; Koziel, V.; Humbert, A.C.; Daval, J.L. Repeated Seizure-Associated Long-Lasting Changes of N-Methyl-D-Aspartate Receptor Properties in the Developing Rat Brain. Int. J. Dev. Neurosci. 1999, 17, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Conn, P.J. Molecular Insights into Metabotropic Glutamate Receptor Allosteric Modulation. Mol. Pharmacol. 2015, 88, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Iacovelli, L.; Di Cicco, G.; Grayson, B.; Rimmer, L.; Fletcher, J.; Neill, J.C.; Wall, M.J.; Ngomba, R.T.; Harte, M. The Comparative Effects of mGlu5 Receptor Positive Allosteric Modulators VU0409551 and VU0360172 on Cognitive Deficits and Signalling in the Sub-Chronic PCP Rat Model for Schizophrenia. Neuropharmacology 2022, 208, 108982. [Google Scholar] [CrossRef]
- Zoicas, I.; Kornhuber, J. The Role of the N-Methyl-D-Aspartate Receptors in Social Behavior in Rodents. Int. J. Mol. Sci. 2019, 20, 5599. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Angelicheva, D.; Vile, D.; Chandler, D.; Morar, B.; Cavanaugh, J.A.; Visscher, P.M.; Jablensky, A.; Pfleger, K.D.; Kalaydjieva, L. Deleterious GRM1 Mutations in Schizophrenia. PLoS ONE 2012, 7, e32849. [Google Scholar] [CrossRef]
- Dogra, S.; Conn, P.J. Metabotropic Glutamate Receptors as Emerging Targets for the Treatment of Schizophrenia. Mol. Pharmacol. 2022, 101, 275–285. [Google Scholar] [CrossRef]
- Wierońska, J.M.; Kusek, M.; Tokarski, K.; Wabno, J.; Froestl, W.; Pilc, A. The GABAB Receptor Agonist CGP44532 and the Positive Modulator GS39783 Reverse Some Behavioural Changes Related to Positive Syndromes of Psychosis in Mice. Br. J. Pharmacol. 2011, 163, 1523–1535. [Google Scholar] [CrossRef]
- Pál, B. Involvement of Extrasynaptic Glutamate in Physiological and Pathophysiological Changes of Neuronal Excitability. Cell. Mol. Life Sci. 2018, 75, 2917–2949. [Google Scholar] [CrossRef]
- Roberts, R.C.; McCollum, L.A.; Schoonover, K.E.; Mabry, S.J.; Roche, J.K.; Lahti, A.C. Ultrastructural Evidence for Glutamatergic Dysregulation in Schizophrenia. Schizophr. Res. 2020, 222, 399–409. [Google Scholar] [CrossRef]
- Benesh, J.L.; Mueller, T.M.; Meador-Woodruff, J.H. AMPA Receptor Subunit Localization in Schizophrenia Anterior Cingulate Cortex. Schizophr. Res. 2022, 249, 16–24. [Google Scholar] [CrossRef]
- McCullumsmith, R.E.; Rowland, L.M. Postmortem, In Silico, and Clinical Studies Focused on Perturbations of Glutamate Neurobiology in Schizophrenia. Schizophr. Res. 2022, 249, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, A.V.; Malkin, S.L.; Postnikova, T.Y.; Smolensky, I.V.; Zubareva, O.E.; Romanova, I.V.; Zakharova, M.V.; Karyakin, V.B.; Zavyalov, V. Ceftriaxone Treatment Affects EAAT2 Expression and Glutamatergic Neurotransmission and Exerts a Weak Anticonvulsant Effect in Young Rats. Int. J. Mol. Sci. 2019, 20, 5852. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.C.K. Current Approaches to Enhance Glutamate Transporter Function and Expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef] [PubMed]
- Abulseoud, O.A.; Alasmari, F.; Hussein, A.M.; Sari, Y. Ceftriaxone as a Novel Therapeutic Agent for Hyperglutamatergic States: Bridging the Gap Between Preclinical Results and Clinical Translation. Front. Neurosci. 2022, 16, 841036. [Google Scholar] [CrossRef]
- Poljak, L.; Miše, B.; Čičin-Šain, L.; Tvrdeić, A. Ceftriaxone Inhibits Conditioned Fear and Compulsive-like Repetitive Marble Digging without Central Nervous System Side Effects Typical of Diazepam—A Study on DBA2/J Mice and a High-5HT Subline of Wistar-Zagreb 5HT Rats. Biomedicines 2024, 12, 1711. [Google Scholar] [CrossRef]
- Research Foundation for Mental Hygiene, Inc. A Placebo-Controlled Efficacy Study of IV Ceftriaxone for Refractory Psychosis. 2009. Available online: https://clinicaltrials.gov/study/NCT00591318 (accessed on 21 January 2025).
- Kamiński, K.; Socała, K.; Abram, M.; Jakubiec, M.; Reeb, K.L.; Temmermand, R.; Zagaja, M.; Maj, M.; Kolasa, M.; Faron-Górecka, A.; et al. Enhancement of Glutamate Uptake as Novel Antiseizure Approach: Preclinical Proof of Concept. Ann. Neurol. 2025, 97, 344–357. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224. [Google Scholar] [CrossRef]
- McQueen, G.; Lally, J.; Collier, T.; Zelaya, F.; Lythgoe, D.J.; Barker, G.J.; Stone, J.M.; McGuire, P.; MacCabe, J.H.; Egerton, A. Effects of N-Acetylcysteine on Brain Glutamate Levels and Resting Perfusion in Schizophrenia. Psychopharmacology 2018, 235, 3045–3054. [Google Scholar] [CrossRef]
- McQueen, G.; Lay, A.; Lally, J.; Gabay, A.S.; Collier, T.; Lythgoe, D.J.; Barker, G.J.; Stone, J.M.; McGuire, P.; MacCabe, J.H.; et al. Effect of Single Dose N-Acetylcysteine Administration on Resting State Functional Connectivity in Schizophrenia. Psychopharmacology 2020, 237, 443–451. [Google Scholar] [CrossRef]
- Bradlow, R.C.J.; Berk, M.; Kalivas, P.W.; Back, S.E.; Kanaan, R.A. The Potential of N-Acetyl-L-Cysteine (NAC) in the Treatment of Psychiatric Disorders. CNS Drugs 2022, 36, 451–482. [Google Scholar] [CrossRef]
- Rossell, S.L.; Francis, P.S.; Galletly, C.; Harris, A.; Siskind, D.; Berk, M.; Bozaoglu, K.; Dark, F.; Dean, O.; Liu, D.; et al. N-Acetylcysteine (NAC) in Schizophrenia Resistant to Clozapine: A Double Blind Randomised Placebo Controlled Trial Targeting Negative Symptoms. BMC Psychiatry 2016, 16, 320. [Google Scholar] [CrossRef] [PubMed]
- Neill, E.; Rossell, S.L.; Yolland, C.; Meyer, D.; Galletly, C.; Harris, A.; Siskind, D.; Berk, M.; Bozaoglu, K.; Dark, F.; et al. N-Acetylcysteine (NAC) in Schizophrenia Resistant to Clozapine: A Double-Blind, Randomized, Placebo-Controlled Trial Targeting Negative Symptoms. Schizophr. Bull. 2022, 48, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Yolland, C.; Hanratty, D.; Neill, E.; Rossell, S.L. Meta-Analysis of Randomised Controlled Trials with N-Acetylcysteine in the Treatment of Schizophrenia. Aust. N. Z. J. Psychiatry 2020, 54, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Rapado-Castro, M.; Dodd, S.; Bush, A.I.; Malhi, G.S.; Skvarc, D.R.; On, Z.X.; Berk, M.; Dean, O.M. Cognitive Effects of Adjunctive N-Acetyl Cysteine in Psychosis. Psychol. Med. 2017, 47, 866–876. [Google Scholar] [CrossRef]
- Klauser, P.; Xin, L.; Fournier, M.; Griffa, A.; Cleusix, M.; Jenni, R.; Cuenod, M.; Gruetter, R.; Hagmann, P.; Conus, P.; et al. N-Acetylcysteine Add-On Treatment Leads to an Improvement of Fornix White Matter Integrity in Early Psychosis: A Double-Blind Randomized Placebo-Controlled Trial. Transl. Psychiatry 2018, 8, 220. [Google Scholar] [CrossRef]
- Lai, C.-C.; Baskaran, R.; Tsao, C.-Y.; Tuan, L.-H.; Siow, P.-F.; Palani, M.; Lee, L.J.-H.; Liu, C.-M.; Hwu, H.-G.; Lee, L.-J. Chronic N-Acetylcysteine Treatment Prevents Amphetamine-Induced Hyperactivity in Heterozygous Disc1 Mutant Mice, a Putative Prodromal Schizophrenia Animal Model. Int. J. Mol. Sci. 2022, 23, 9419. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The Role of Glutamate Transporters in the Pathophysiology of Neuropsychiatric Disorders. NPJ Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef]
- Deakin, B.; Suckling, J.; Barnes, T.R.E.; Byrne, K.; Chaudhry, I.B.; Dazzan, P.; Drake, R.J.; Giordano, A.; Husain, N.; Jones, P.B.; et al. The Benefit of Minocycline on Negative Symptoms of Schizophrenia in Patients with Recent-Onset Psychosis (BeneMin): A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Psychiatry 2018, 5, 885–894. [Google Scholar] [CrossRef]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, Expression, and Function of Kynurenine Aminotransferases in Human and Rodent Brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef]
- Kynexis. A Study to Investigate the Safety, Tolerability, and Pharmacokinetics of KYN-5356 in Healthy Subjects Aged 18 to 55 Years. 2024. Available online: https://clinicaltrials.gov/study/NCT06225115 (accessed on 18 February 2025).
- Royo, M.; Escolano, B.A.; Madrigal, M.P.; Jurado, S. AMPA Receptor Function in Hypothalamic Synapses. Front. Synaptic Neurosci. 2022, 14, 833449. [Google Scholar] [CrossRef]
- Zhang, H.; Bramham, C.R. Bidirectional Dysregulation of AMPA Receptor-Mediated Synaptic Transmission and Plasticity in Brain Disorders. Front. Synaptic Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Kadriu, A.; Musazzi, L.; Johnston, J.N.; Kalynchuk, L.E.; Caruncho, H.J.; Popoli, M.; Zarate, C.A. Positive AMPA Receptor Modulation in the Treatment of Neuropsychiatric Disorders: A Long and Winding Road. Drug Discov. Today 2021, 26, 2816–2838. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lamberti, J.S.; Leon, A.C.; Green, M.F.; Miller, A.L.; Patel, J.; Manschreck, T.; Freudenreich, O.; Johnson, S.A. A Placebo-Controlled Add-On Trial of the Ampakine, CX516, for Cognitive Deficits in Schizophrenia. Neuropsychopharmacology 2008, 33, 465–472. [Google Scholar] [CrossRef]
- Witkin, J.M.; Li, G.; Golani, L.K.; Xiong, W.; Smith, J.L.; Ping, X.; Rashid, F.; Jahan, R.; Cerne, R.; Cook, J.M.; et al. The Positive Allosteric Modulator of α2/3-Containing GABAA Receptors, KRM-II-81, Is Active in Pharmaco-Resistant Models of Epilepsy and Reduces Hyperexcitability after Traumatic Brain Injury. J. Pharmacol. Exp. Ther. 2020, 372, 83–94. [Google Scholar] [CrossRef]
- Sharma, R.; Nakamura, M.; Neupane, C.; Jeon, B.H.; Shin, H.; Melnick, S.M.; Glenn, K.J.; Jang, I.-S.; Park, J.B. Positive Allosteric Modulation of GABAA Receptors by a Novel Antiepileptic Drug Cenobamate. Eur. J. Pharmacol. 2020, 879, 173117. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin. J. Neuroendocrinol. 2021, 33, e12934. [Google Scholar] [CrossRef]
- Etchecopar-Etchart, D.; Yon, D.K.; Wojciechowski, P.; Aballea, S.; Toumi, M.; Boyer, L.; Fond, G. Comprehensive Evaluation of 45 Augmentation Drugs for Schizophrenia: A Network Meta-Analysis. EClinicalMedicine 2024, 69, 102473. [Google Scholar] [CrossRef]
- Zhu, E.; Mathew, D.; Jee, H.J.; Sun, M.; Liu, W.; Zhang, Q.; Wang, J. AMPAkines Have Site-Specific Analgesic Effects in the Cortex. Mol. Pain 2024, 20, 17448069231214677. [Google Scholar] [CrossRef]

{kind=link}
| Study | Findings | Implications |
|---|---|---|
| Merritt et al. [20] | Meta-analysis found evidence of glutamatergic elevations in schizophrenia. | Supports the NMDA receptor hypofunction/disinhibition model of schizophrenia. |
| Bojesen et al. [25], Javitt et al. [22], Rowland et al. [26], Stone et al. [27] | Increases in cortical glutamate observed in NMDAR hypofunction produced by ketamine infusion. | Suggests that ketamine-induced glutamate increase can model aspects of schizophrenia. |
| Wenneberg et al. [28] | No overall difference in medial frontal Glx levels in high-risk individuals compared with controls. | Challenges previous findings of elevated glutamate in high-risk individuals. |
| de la Fuente-Sandoval et al. [23] | Elevated glutamate levels in the striatum in at-risk individuals who later transition to psychosis. | Early biomarker for individuals transitioning to psychosis. |
| Bossong et al. [24] | Elevated hippocampal glutamate levels in at-risk individuals who later transition to psychosis. | Further supports glutamate as a predictive factor for psychosis transition. |
| Egerton et al. [29] | Lower thalamic glutamate levels associated with continued presence of attenuated symptoms at follow-up. | Implicates thalamic glutamate levels in symptom persistence. |
| Demjaha et al. [30], Egerton et al. [31], Iwata et al. [32], Mouchlianitis et al. [33], Tarumi et al. [34] | Elevated glutamate or Glx in the ACC in patients with non-remission, antipsychotic resistance, or clozapine resistance. | Identifies a subgroup of patients with high glutamate levels who exibit treatment resistance. |
| Goldstein et al. [35] | No observed association between glutamate levels and treatment response in all studies. | Highlights inconsistencies in findings regarding glutamate and treatment response. |
| Egerton et al. [36] | Higher ACC glutamate levels at illness onset associated with higher likelihood of non-remission after treatment. | Glutamate levels are a predictor of treatment response in early psychosis. |
| Merritt et al. [37] | Non-remission associated with increases in Glx in the thalamus over 9 months. | Longitudinal changes in glutamate indicate persistent symptoms. |
| Jelen et al. [38] | Blunted activation of dynamic glutamate responses in ACC to cognitive task in schizophrenia and bipolar disorder. | Suggests schizophrenia involves an impaired glutamate response to cognitive demand. |
| Taylor et al. [39] | Blunted activation of dynamic glutamate responses in ACC to Stroop task in schizophrenia and major depressive disorder. | Identifiesblunted glutamate response as a transdiagnostic feature across psychiatric disorders. |
| Treatment Approach | Examples of Agents | Mechanism of Action | Potential Benefits | Challenges and Limitations |
|---|---|---|---|---|
| NMDA Receptor Modulators | D-serine, Glycine, Bitopertin (GlyT1 inhibitor), Rapastinel | Enhances NMDA receptor activity through co-agonists or glycine transport inhibition | Improves cognitive deficits and negative symptoms [42,43] | Variable efficacy, possible excitotoxicity risks, inconsistent trial results [44] |
| Metabotropic Glutamate Receptor (mGluR) Agents | Pomaglumetad, TS-134, JNJ-40411813 (mGlu2 PAM), AZD8529 | Regulates glutamate transmission via metabotropic receptors | Potentially reduces psychotic symptoms and cognitive impairment [45,46] | Some agents failed in clinical trials, patient variability in response [47] |
| Glutamate Transporter Regulators | Ceftriaxone (EAAT2 upregulation), N-acetylcysteine (NAC) | Enhances glutamate clearance and homeostasis | Restores glutamate balance and prevents excitotoxicity | Limited human trials, difficulty translating preclinical success [48] |
| Kynurenine Pathway Inhibitors | KYN-5356 (KAT II inhibitor) | Reduces kynurenic acid levels to enhance NMDA function | Enhances NMDA function and cognitive processing | Potential side effects, needs further clinical validation [49] |
| AMPA Receptor Modulators | CX516 (Ampakine), Other AMPA-positive allosteric modulators | Enhances AMPA receptor-mediated synaptic transmission | Improves cognitive function and learning processes [50] | Limited evidence, inconsistent trial results |
| Neuroinflammatory modulation | Minocycline, NSAIDs, TNF-alpha inhibitors | Reduces neuroinflammation that disrupts glutamate signaling | May reduce neurotoxicity and cognitive decline | May not directly improve schizophrenia symptoms, mixed efficacy [51] |
| Synaptic Plasticity Enhancers | Intranasal insulin, IGF-1 analogs, BDNF enhancers | Promotes synaptic repair and neuroplasticity | Facilitates recovery by strengthening synaptic connections | Still in early research phase, needs larger clinical trials [52,53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadgyas-Stanculete, M.; Capatina, O.O. Glutamate-Based Therapeutic Strategies for Schizophrenia: Emerging Approaches Beyond Dopamine. Int. J. Mol. Sci. 2025, 26, 4331. https://doi.org/10.3390/ijms26094331
Fadgyas-Stanculete M, Capatina OO. Glutamate-Based Therapeutic Strategies for Schizophrenia: Emerging Approaches Beyond Dopamine. International Journal of Molecular Sciences. 2025; 26(9):4331. https://doi.org/10.3390/ijms26094331
Chicago/Turabian StyleFadgyas-Stanculete, Mihaela, and Octavia Oana Capatina. 2025. "Glutamate-Based Therapeutic Strategies for Schizophrenia: Emerging Approaches Beyond Dopamine" International Journal of Molecular Sciences 26, no. 9: 4331. https://doi.org/10.3390/ijms26094331
APA StyleFadgyas-Stanculete, M., & Capatina, O. O. (2025). Glutamate-Based Therapeutic Strategies for Schizophrenia: Emerging Approaches Beyond Dopamine. International Journal of Molecular Sciences, 26(9), 4331. https://doi.org/10.3390/ijms26094331