
Repercussions of the Calpain Cleavage-Related Missense Mutations in the Cytosolic Domains of Human Integrin-β Subunits on the Calpain–Integrin Signaling Axis


Abstract
1. Introduction
2. Results
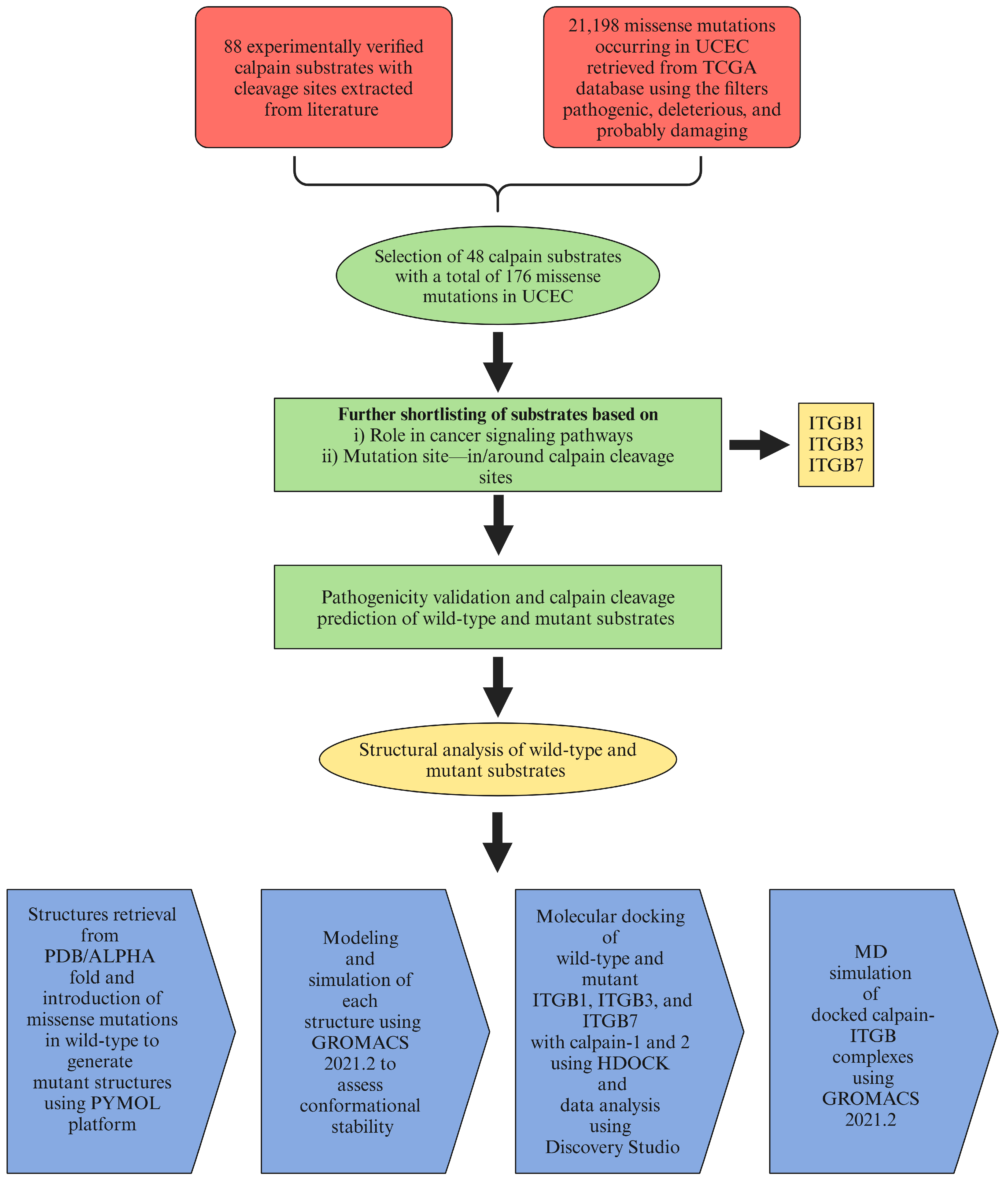
2.1. Data Retrieval
2.2. Effects of Mutations on Pathogenicity and Calpain Cleavage
2.3. Modeling and Simulation
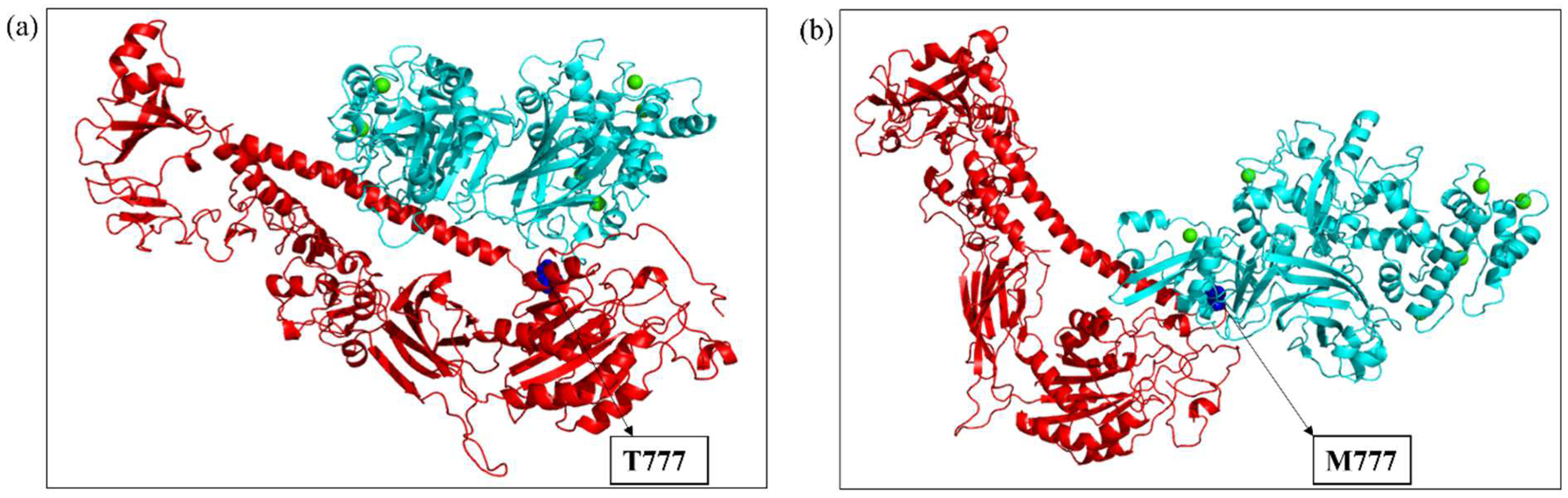
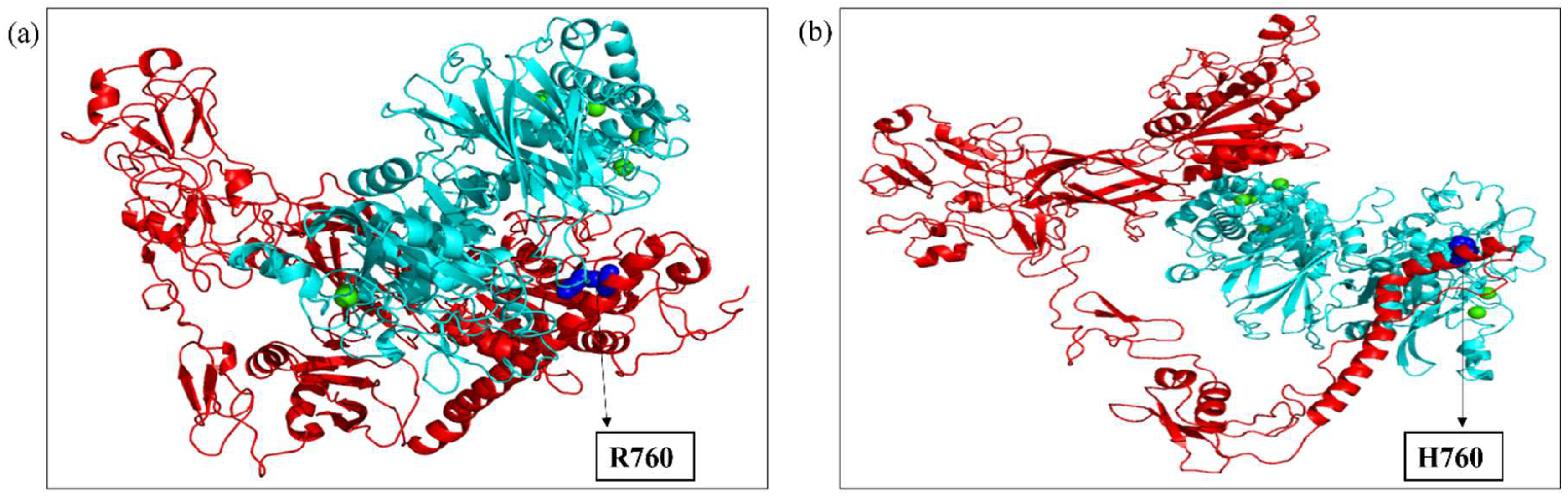
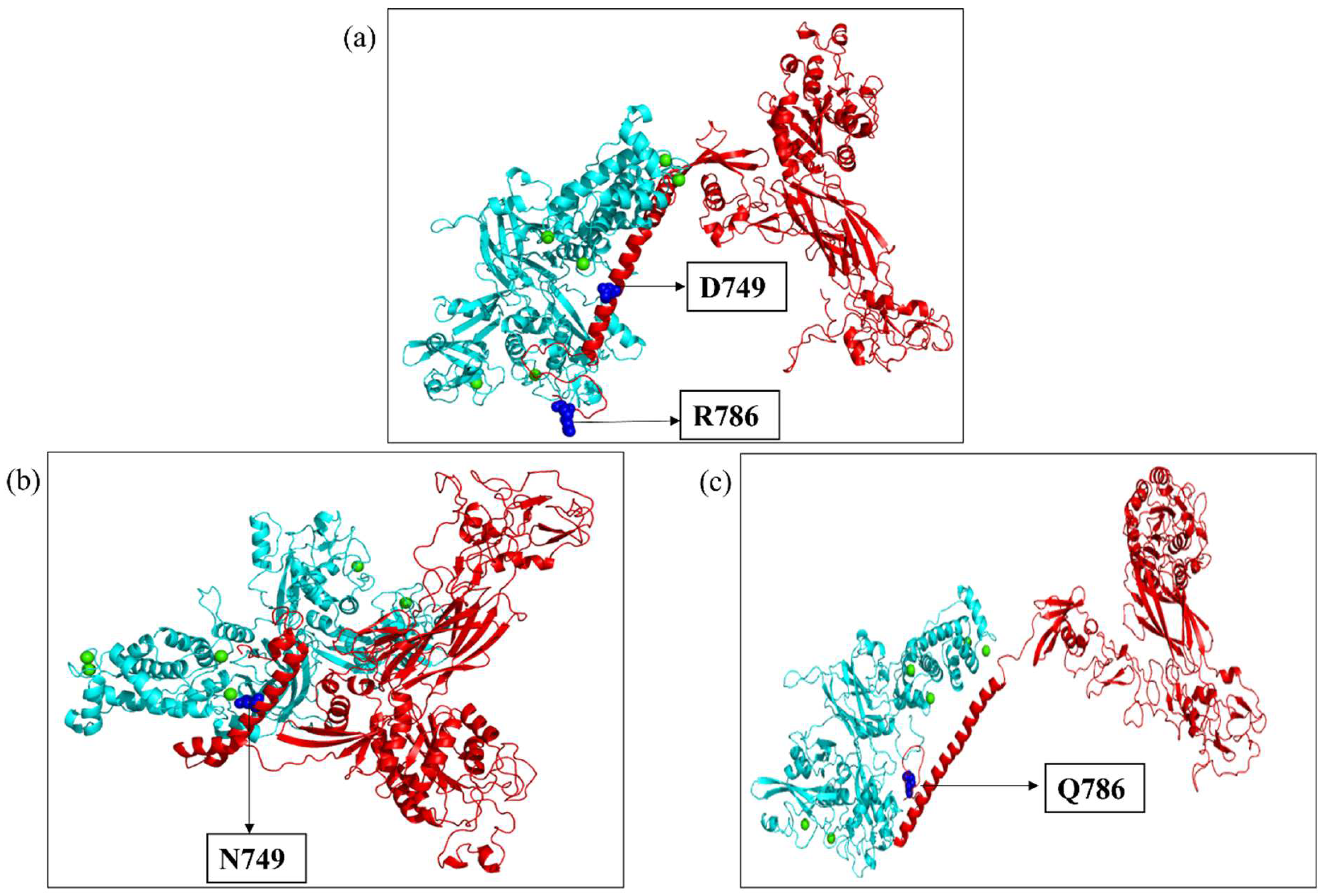
2.4. Molecular Docking
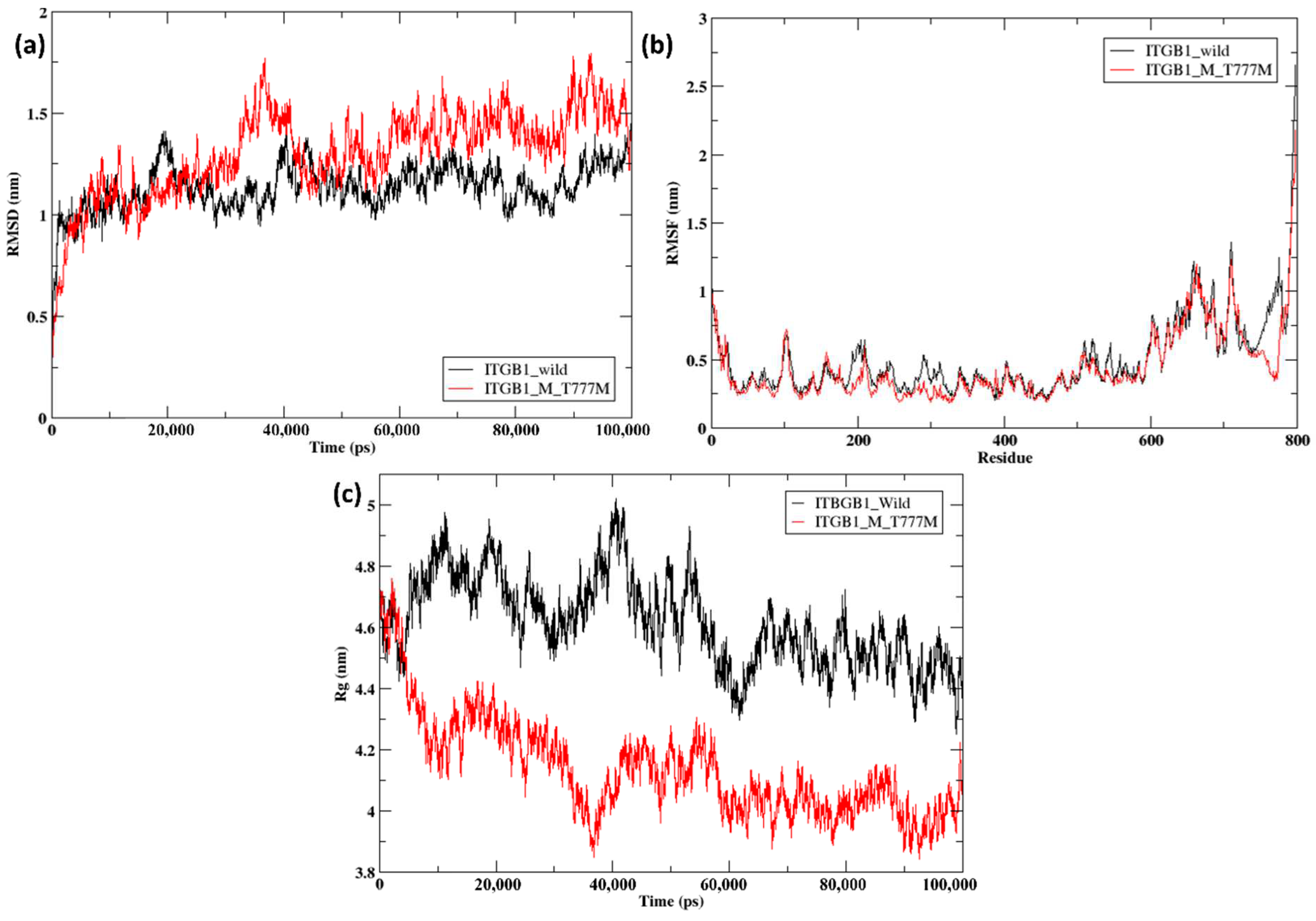
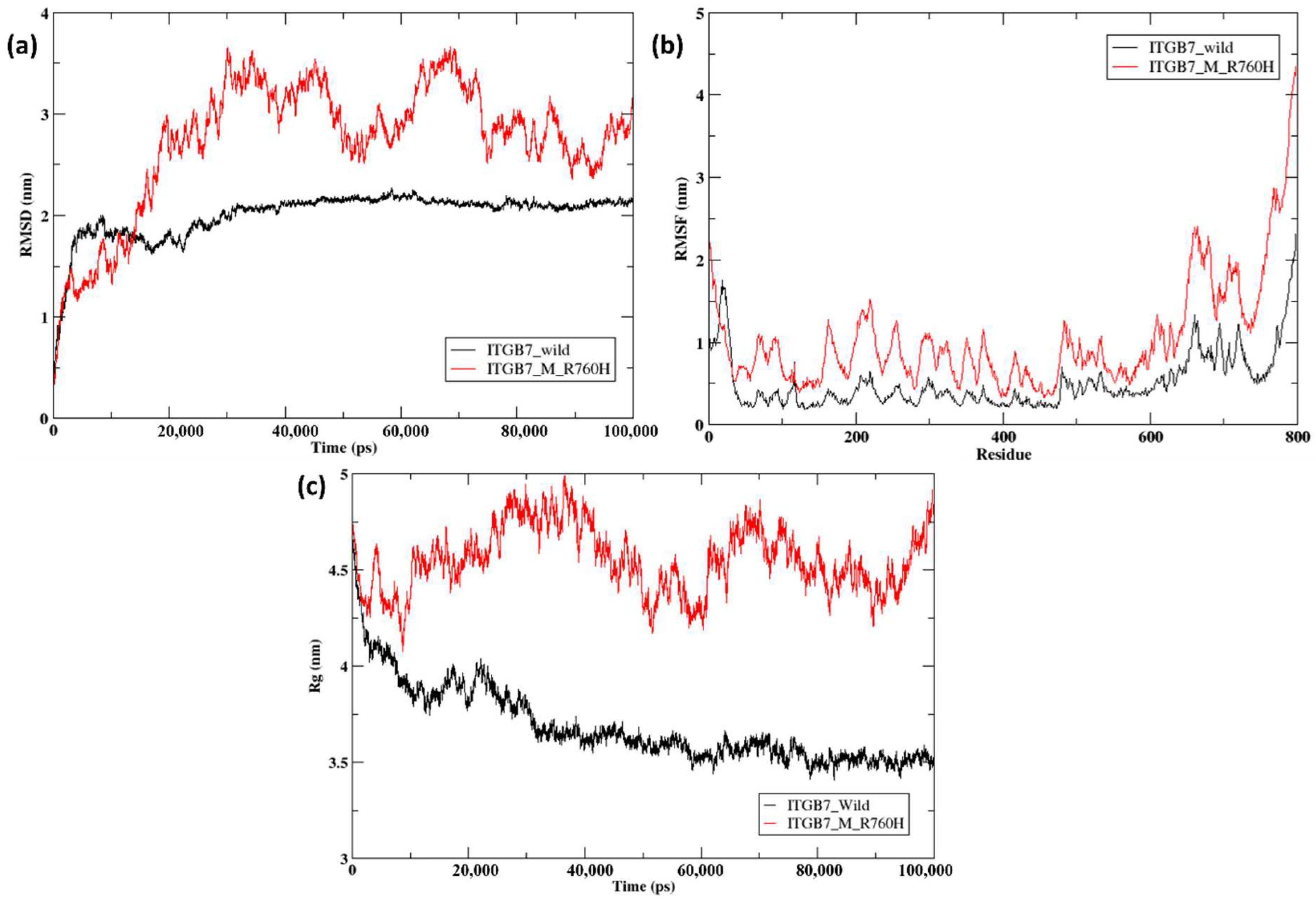
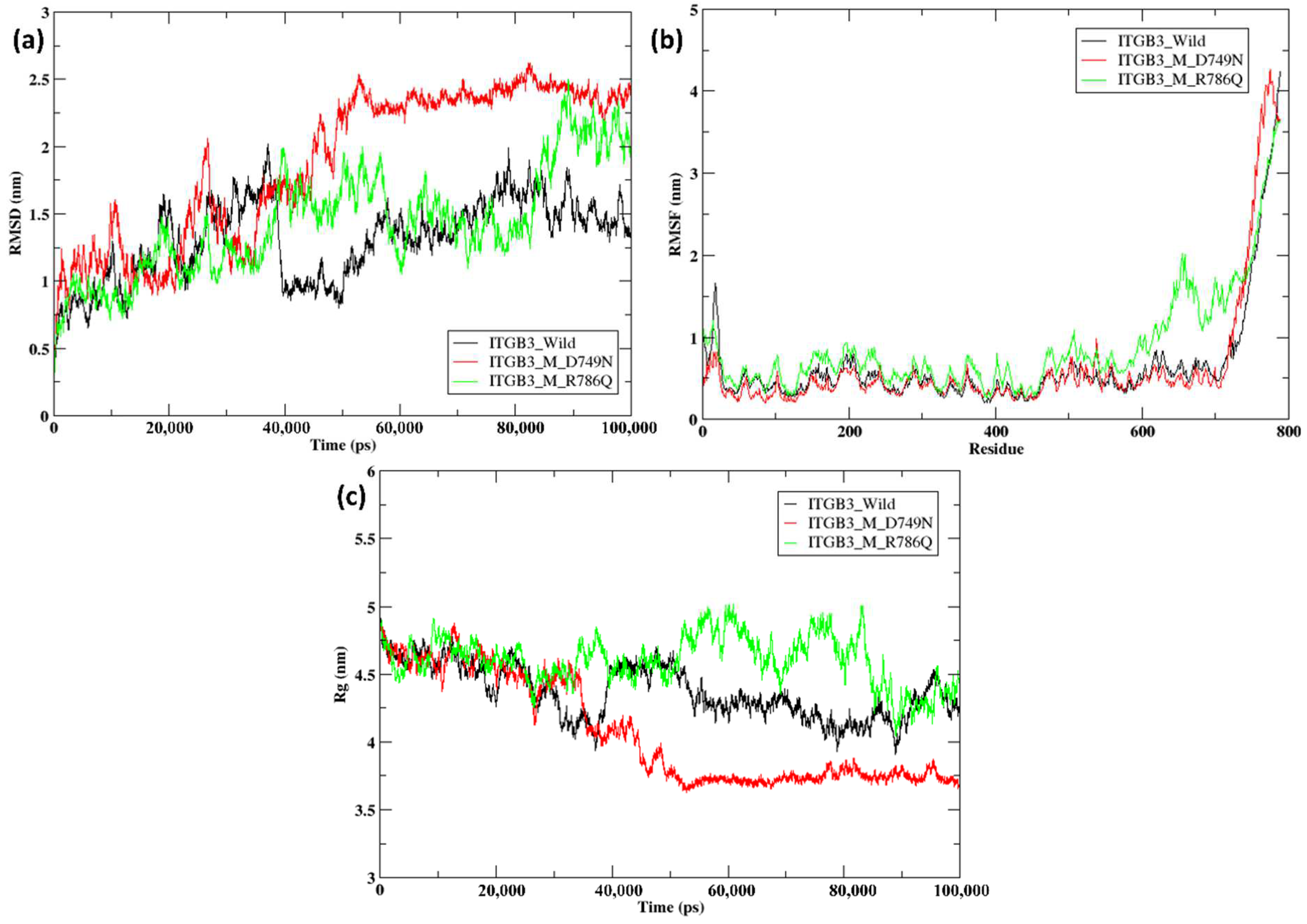
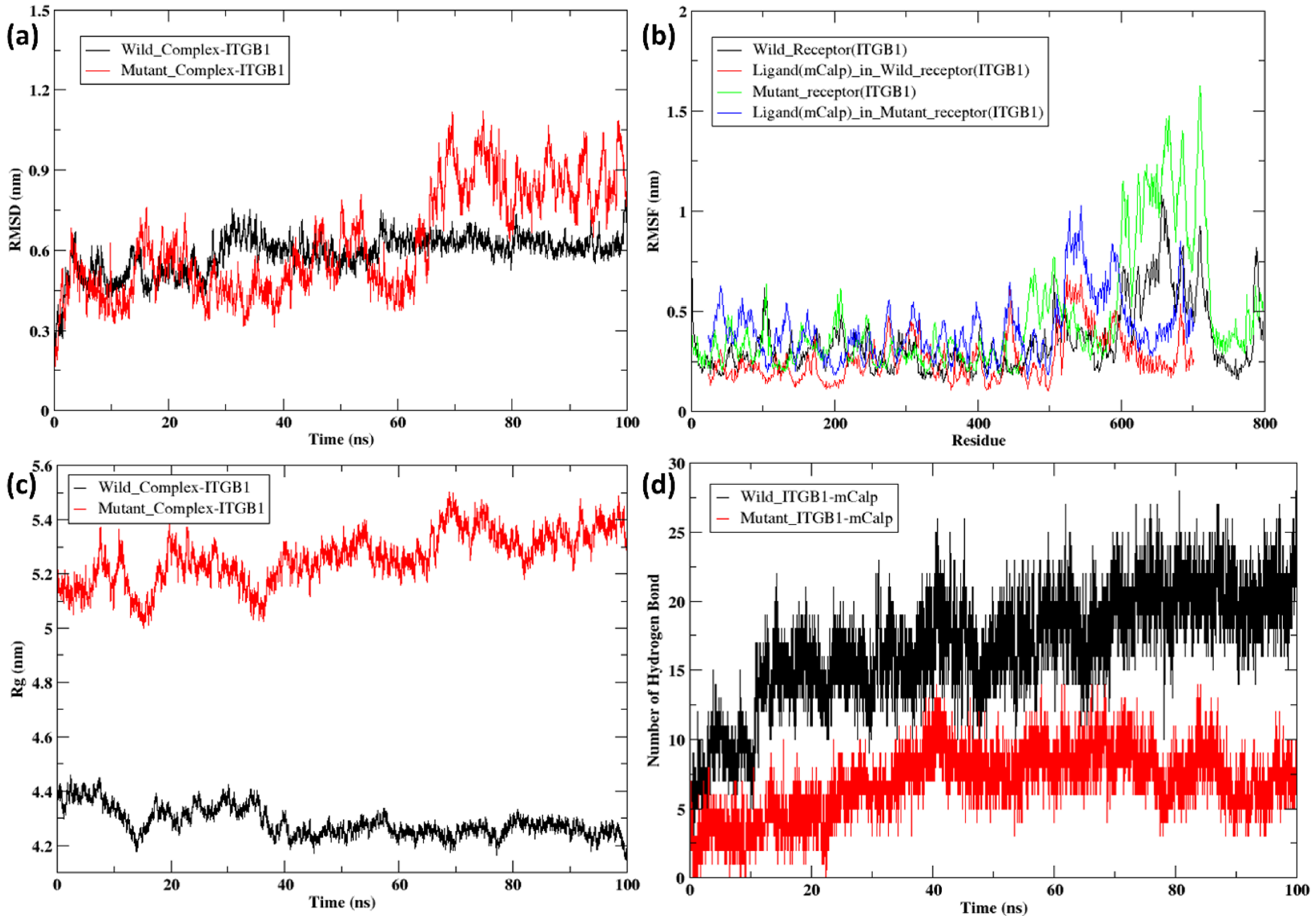
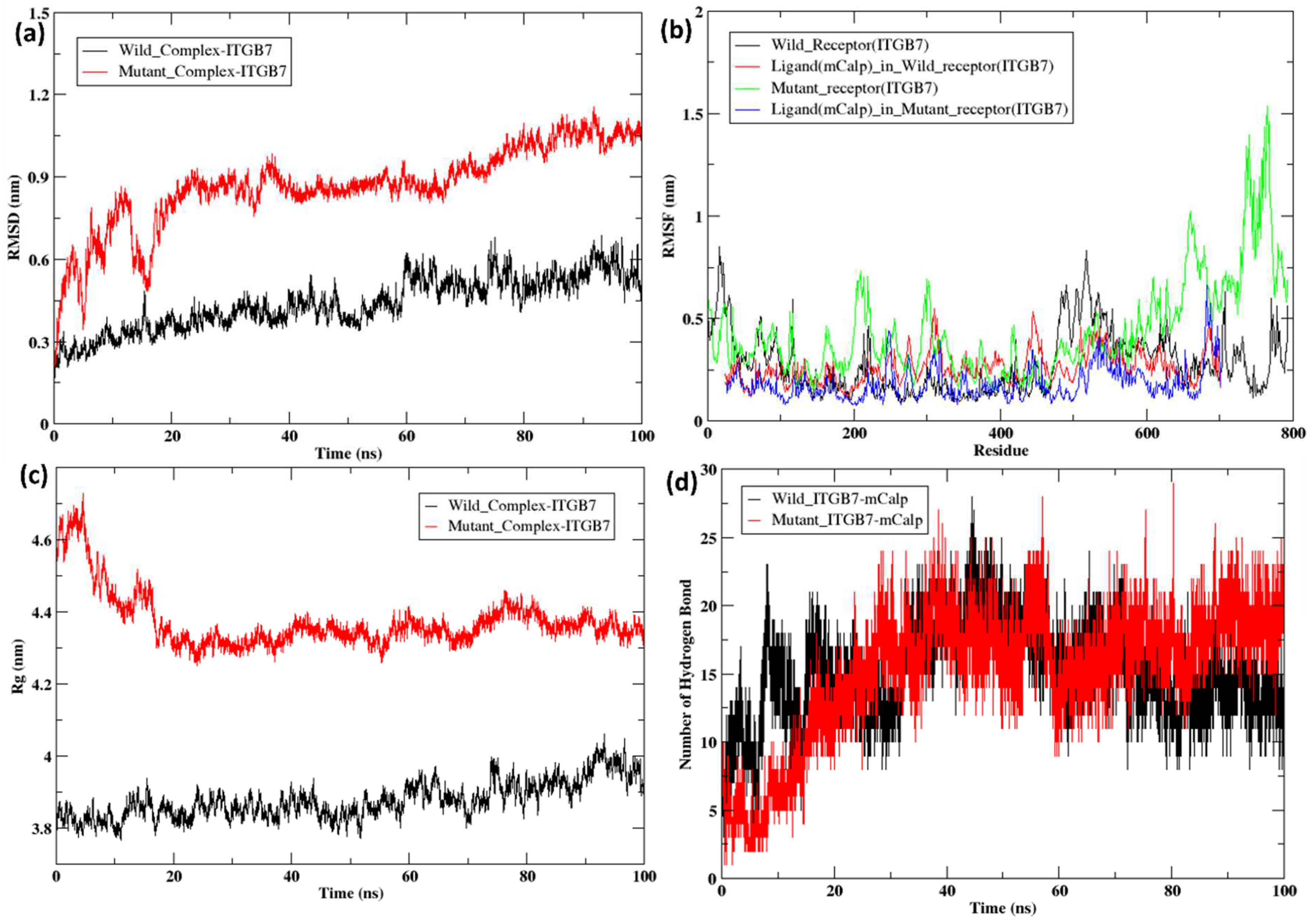
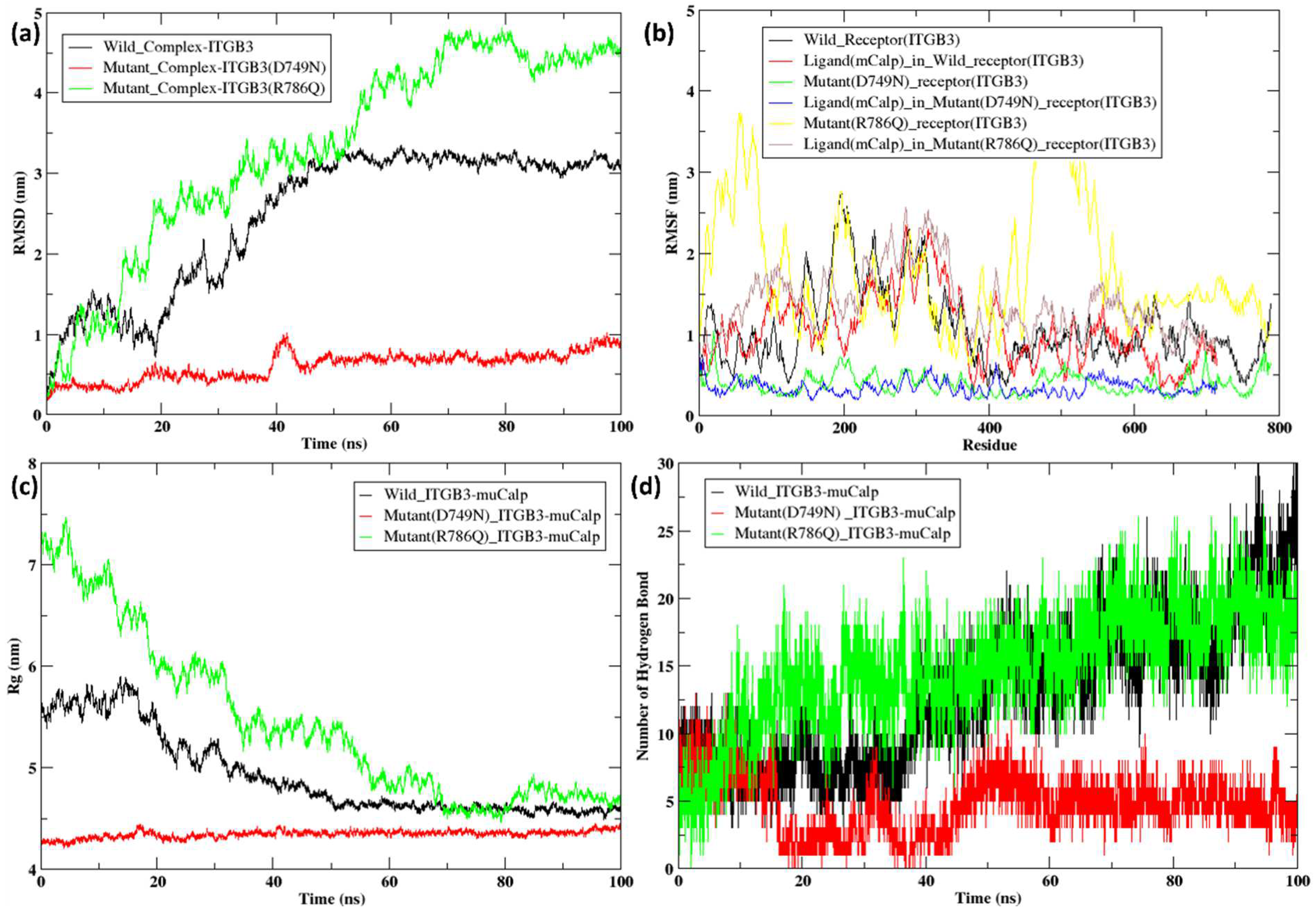
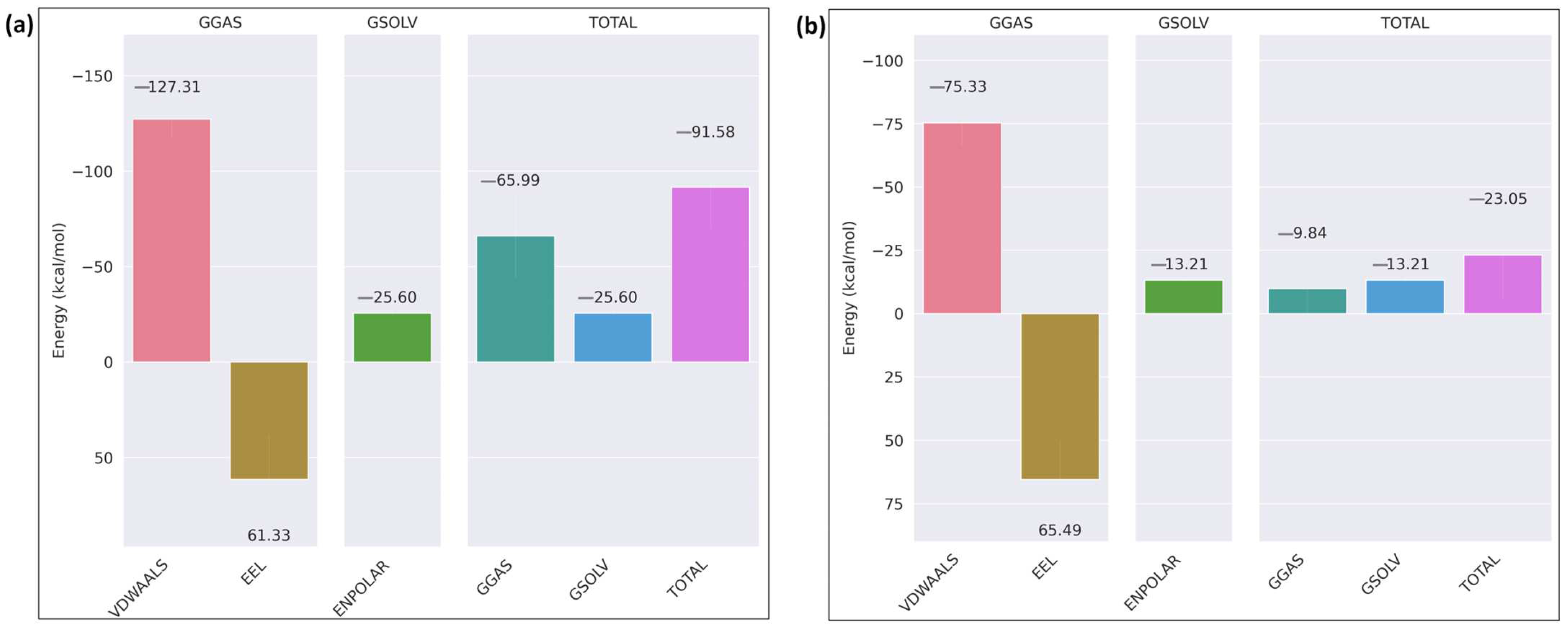
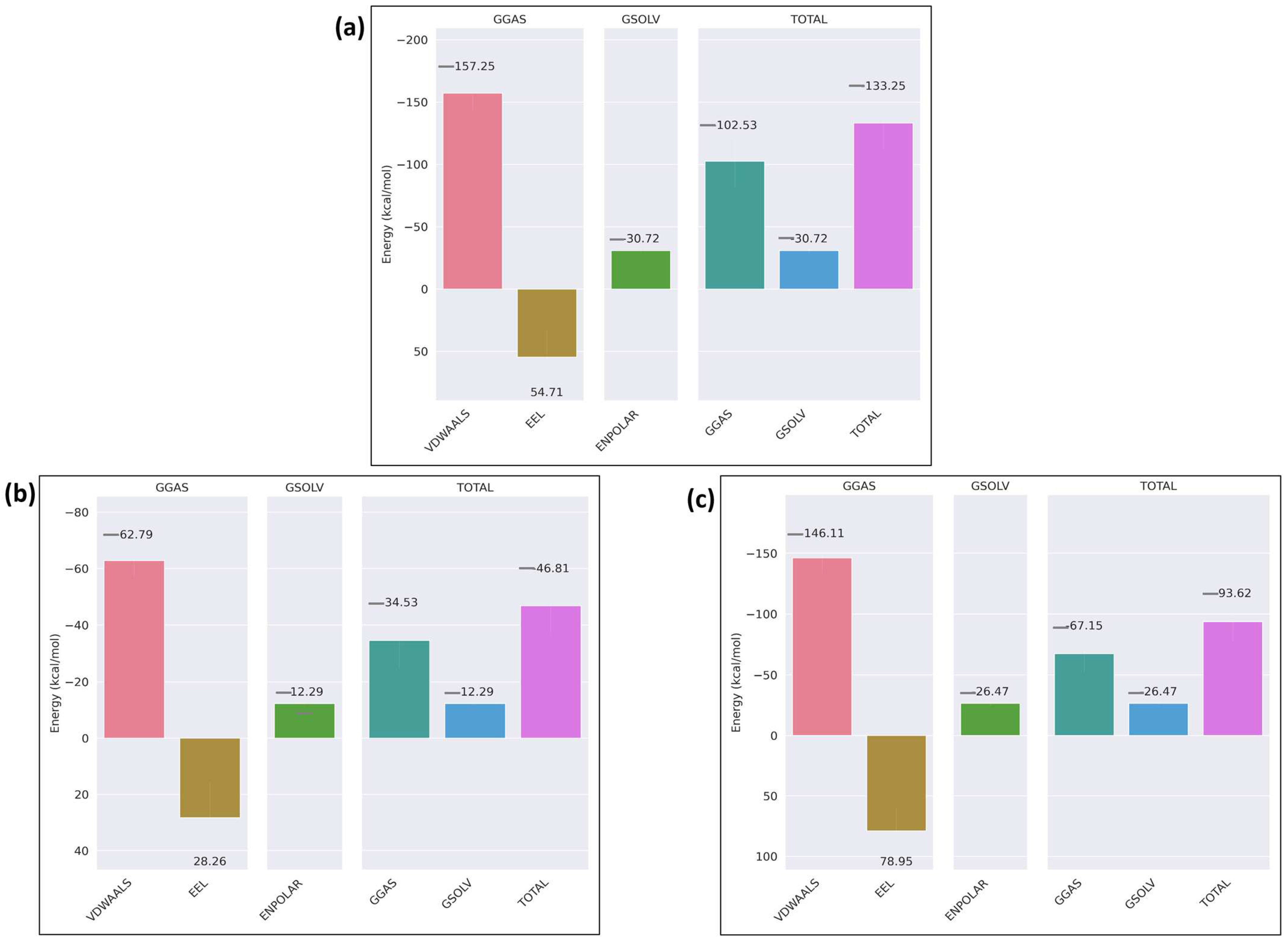
2.5. Molecular Dynamics Simulation of the Docked Complexes
3. Discussion
4. Materials and Methods
4.1. Data Retrieval and Curation
4.2. Pathogenicity Prediction
4.2.1. PolyPhen-2
4.2.2. Fathmm
4.2.3. Panther-PSEP (Protein Analysis Through Evolutionary Relationships)
4.3. Calpain Cleavage Prediction
4.4. Structure Retrieval
4.5. Modeling and Simulation
4.6. Molecular Docking and MD Simulation of Docked Complexes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCRM | calpain cleavage-related mutations |
| UCEC | uterine corpus endometrial carcinoma |
| ITG-β | integrin-β subunits |
| TCGA-GDC | The Cancer Genome Atlas-Genomic Data Commons |
| RMSD | root mean square deviation |
| RMSF | root mean square fluctuation |
References
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bodeau, A.L.; Berrier, A.L.; Mastrangelo, A.M.; Martinez, R.; LaFlamme, S.E. A functional comparison of mutations in integrin β cytoplasmic domains. J. Cell Sci. 2001, 114, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Borgohain, G.; Dan, N.; Paul, S. Use of molecular dynamics simulation to explore structural facets of human prion protein with pathogenic mutations. Biophys. Chem. 2016, 213, 32–39. [Google Scholar] [CrossRef]
- Cai, X.; Thinn AM, M.; Wang, Z.; Shan, H.; Zhu, J. The importance of N-glycosylation on β3 integrin ligand binding and conformational regulation. Sci. Rep. 2017, 7, 4656. [Google Scholar] [CrossRef]
- Carragher, N.O. Calpain Inhibition: A Therapeutic Strategy Targeting Multiple Disease States. Curr. Pharm. Des. 2006, 12, 615–638. [Google Scholar] [CrossRef] [PubMed]
- Rios-Doria, J.; Day, K.C.; Kuefer, R.; Rashid, M.G.; Chinnaiyan, A.M.; Rubin, M.A.; Day, M.L. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J. Biol. Chem. 2003, 278, 1372–1379. [Google Scholar] [CrossRef]
- Storr, S.J.; Carragher, N.O.; Frame, M.C.; Parr, T.; Martin, S.G. The calpain system and cancer. Nat. Rev. Cancer 2011, 11, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Perrin, B.J.; Huttenlocher, A. Calpain. Int. J. Biochem. Cell Biol. 2002, 34, 722–725. [Google Scholar] [CrossRef]
- Chen, L.; Xiao, D.; Tang, F.; Gao, H.; Li, X. CAPN6 in disease: An emerging therapeutic target (Review). Int. J. Mol. Med. 2020, 46, 1644–1652. [Google Scholar] [CrossRef]
- Dasgupta, S.; Sirisha PV, S.; Neelaveni, K.; Anuradha, K.; Reddy, B.M. Association of capn10 snps and haplotypes with polycystic ovary syndrome among south indian women. PLoS ONE 2012, 7, e32192. [Google Scholar] [CrossRef]
- Du, X.; Saido, T.C.; Tsubuki, S.; Indig, F.E.; Williams, M.J.; Ginsberg, M.H. Calpain cleavage of the cytoplasmic domain of the integrin β3 subunit. J. Biol. Chem. 1995, 270, 26146–26151. [Google Scholar] [CrossRef] [PubMed]
- Ennes-Vidal, V.; Menna-Barreto, R.F.S.; Branquinha, M.H.; Dos Santos, A.L.S.; D’Avila-Levy, C.M. Why calpain inhibitors are interesting leading compounds to search for new therapeutic options to treat leishmaniasis? Parasitology 2017, 144, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Fujiwara, H.; Morita, K.; Watt, F.M. An activating β1 integrin mutation increases the conversion of benign to malignant skin tumors. Cancer Res. 2009, 69, 1334–1342. [Google Scholar] [CrossRef]
- Yadavalli, R.; Guttmann, R.P.; Seward, T.; Centers, A.P.; Williamson, R.A.; Telling, G.C. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J. Biol. Chem. 2004, 279, 21948–21956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Deen, S.; Storr, S.J.; Chondrou, P.S.; Nicholls, H.; Yao, A.; Rungsakaolert, P.; Martin, S.G. Calpain system protein expression and activity in ovarian cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 345–361. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Taylor, R.G.; Ouali, A. The calpain system and skeletal muscle growth. Can. J. Anim. Sci. 1998, 78, 503–512. [Google Scholar] [CrossRef]
- Guyon, J.R.; Kudryashova, E.; Potts, A.; Dalkilic, I.; Brosius, M.A.; Thompson, T.G.; Beckmann, J.S.; Kunkel, L.M.; Spencer, M.J. Calpain 3 cleaves filamin C and regulates its ability to interact with-and-sarcoglycans. Muscle Nerve 2003, 28, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Hosfield, C.M.; Moldoveanu, T.; Davies, P.L.; Elce, J.S.; Jia, Z. Calpain Mutants with Increased Ca2+ Sensitivity and Implications for the Role of the C2-like Domain. J. Biol. Chem. 2001, 276, 7404–7407. [Google Scholar] [CrossRef]
- Howe, G.A.; Addison, C.L. β1 integrin: An emerging player in the modulation of tumorigenesis and response to therapy. Cell Adhes. Migr. 2012, 6, 71–77. [Google Scholar] [CrossRef]
- Huang, X.-Z.; Chen, A.; Agrez, M.; Sheppard, D. A Point Mutation in the Integrin {36 Subunit Abolishes Both exv{36 Binding to Fibronectin and Receptor Localization to Focal Contacts. Am. J. Respir. Cell Mol. Biol. 1995, 13, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Petrounevitch, V.; Wong, A.; Moldoveanu, T.; Davies, P.L.; Elce, J.S.; Beckmann, J.S. Mutations in calpain 3 associated with limb girdle muscular dystrophy: Analysis by molecular modeling and by mutation in m-calpain. Biophys. J. 2001, 80, 2590–2596. [Google Scholar] [CrossRef] [PubMed]
- Penna, I.; Du, H.; Ferriani, R.; Taylor, H.S. Calpain5 expression is decreased in endometriosis and regulated by HOXA10 in human endometrial cells. Mol. Hum. Reprod. 2008, 14, 613–618. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kovacheva, M.; Zepp, M.; Berger, S.; Berger, M.R. Conditional knockdown of integrin beta-3 reveals its involvement in osteolytic and soft tissue lesions of breast cancer skeletal metastasis. J. Cancer Res. Clin. Oncol. 2021, 147, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Pocheć, E.; Lityńska, A.; Amoresano, A.; Casbarra, A. Glycosylation profile of integrin α3β1 changes with melanoma progression. Biochim. et Biophys. Acta–Mol. Cell Res. 2003, 1643, 113–123. [Google Scholar] [CrossRef]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef]
- Leloup, L.; Wells, A. Calpains as potential anti-cancer targets. Expert Opin. Ther. Targets 2011, 15, 309–323. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System, Version 1.3r1; Schrödinger, Inc.: New York, NY, USA, 2010.
- Liu, Z.X.; Yu, K.; Dong, J.; Zhao, L.; Liu, Z.; Zhang, Q.; Li, S.; Du, Y.; Cheng, H. Precise prediction of calpain cleavage sites and their aberrance caused by mutations in cancer. Front. Genet. 2019, 10, 715. [Google Scholar] [CrossRef]
- Tompa, P.; Buzder-Lantos, P.; Tantos, A.; Farkas, A.; Szilágyi, A.; Bánóczi, Z.; Hudecz, F.; Friedrich, P. On the sequential determinants of calpain cleavage. J. Biol. Chem. 2004, 279, 20775–20785. [Google Scholar] [CrossRef] [PubMed]
- Pariat, M.; Carillo, S.; Molinari, M.; Salvat, C.; Debüssche, L.; Debüssche, D.; Bracco, L.; Milner, J.O.; Piechaczyk, M. Proteolysis by Calpains: A Possible Contribution to Degradation of p53. Mol. Cell. Biol. 1997, 17, 2806–2815. [Google Scholar] [CrossRef]
- Singh, B.; Bulusu, G.; Mitra, A. Effects of point mutations on the thermostability of B. subtilis lipase: Investigating nonadditivity. J. Comput. Mol. Des. 2016, 30, 899–916. [Google Scholar] [CrossRef]
- Rucker, G.; Qin, H.; Zhang, L. Structure, dynamics and free energy studies on the effect of point mutations on SARS-CoV-2 spike protein binding with ACE2 receptor. PLoS ONE 2023, 18, e0289432. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, T.; Pei, J.; Ouyang, Q. Mutation-induced protein interaction kinetics changes affect apoptotic network dynamic properties and facilitate oncogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E4046–E4054. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ye, Y.; Sang, P.; Yin, Y.; Hu, W.; Wang, J.; Zhang, C.; Li, D.; Wan, W.; Li, R.; et al. Effect of R119G Mutation on Human P5CR1 Dynamic Property and Enzymatic Activity. BioMed Res. Int. 2017, 2017, 4184106. [Google Scholar] [CrossRef]
- Nagasundaram, N.; Zhu, H.; Liu, J.; Karthick, V.; George Priya Doss, C.; Chakraborty, C.; Chen, L. Analysing the effect of mutation on protein function and discovering potential inhibitors of CDK4: Molecular modelling and dynamics studies. PLoS ONE 2015, 10, e0133969. [Google Scholar] [CrossRef]
- Kubbutat, M.H.G.; Vousden, K.H. Proteolytic Cleavage of Human p53 by Calpain: A Potential Regulator of Protein Stability. Mol. Cell. Biol. 1997, 17, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, P.; Shattil, S.J.; Ablooglu, A.J. C-terminal COOH of integrin β1 is necessary for β1 association with the kindlin-2 adapter protein. J. Biol. Chem. 2014, 289, 11183–11193. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, M.; Du, X.; Ginsberg, M.H. Calpain cleavage of integrin β cytoplasmic domains. FEBS Lett. 1999, 460, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ylänne, J.; Huuskonen, J.; O’Toole, T.E.; Ginsberg, M.H.; Virtanen, I.; Gahmberg, C.G. Mutation of the Cytoplasmic Domain of the Integrin β3 Subunit: Differential effects on cell spreading, recruitment to adhesion plaques, endocytosis, and phagocytosis. J. Biol. Chem. 1995, 270, 9550–9557. [Google Scholar] [CrossRef]
- Qiu, Z.; Sheesley, P.; Ahn, J.H.; Yu, E.J.; Lee, M. A novel mutation in an NPXY motif of β integrin reveals phenotypes similar to him-4/hemicentin. Front. Cell Dev. Biol. 2019, 7, 247. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef]
- Shapovalov, I.; Harper, D.; Greer, P.A. Calpain as a therapeutic target in cancer. Expert Opin. Ther. Targets 2022, 26, 217–231. [Google Scholar] [CrossRef]
- Sharma, J.; Mulherkar, S.; Chen, U.I.; Xiong, Y.; Bajaj, L.; Cho, B.K.; Goo, Y.A.; Leung, H.C.E.; Tolias, K.F.; Sardiello, M. Calpain activity is negatively regulated by a KCTD7–Cullin-3 complex via non-degradative ubiquitination. Cell Discov. 2023, 9, 32. [Google Scholar] [CrossRef]
- Shiraha, H.; Glading, A.; Chou, J.; Jia, Z.; Wells, A. Activation of m-Calpain (Calpain II) by Epidermal Growth Factor Is Limited by Protein Kinase A Phosphorylation of m-Calpain. Mol. Cell. Biol. 2002, 22, 2716–2727. [Google Scholar] [CrossRef]
- Boekhorst, V.T.; Jiang, L.; Mählen, M.; Meerlo, M.; Dunkel, G.; Durst, F.C.; Yang, Y.; Levine, H.; Burgering, B.M.T.; Friedl, P. Calpain-2 regulates hypoxia/HIF-induced plasticity toward amoeboid cancer cell migration and metastasis. Curr. Biol. 2022, 32, 412–427.e8. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Sim (Ana) Analogue Release 2024, Growdea Technologies Pvt. Lt. 2024; v1.1. Available online: https://growdeatech.com/Analogue/ (accessed on 23 August 2024).
- Trajecta (Ana) Analogue Release 2024, Growdea Technologies Pvt. Lt. 2024; v1.1. Available online: https://growdeatech.com/Analogue/ (accessed on 23 August 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Mutations | Polyphen2 (0–1) | Fathmm (−0.75) | Panther |
|---|---|---|---|---|
| ITGB1 | T777M | Most likely damaging (0.998) | Cancer (−1.03) | Damaging (0.316) |
| ITGB7 | R760H | Most likely damaging (1.00) | Cancer (−1.31) | Damaging (0.280) |
| ITGB3 | D749N | Most likely damaging (1.00) | Cancer (−0.98) | Most likely damaging (0.85) |
| R786Q | Most likely damaging (1.00) | Cancer (−0.98) | Most likely damaging (0.57) |
| Substrates | Site of Mutation | Total Number of Calpain Cleavage Sites Predicted | Calpain Cleavage Site Variation Around Mutation Site | |
|---|---|---|---|---|
| Wild-Type | Mutant | |||
| ITGB1 | T777M | Wild-type: 05 Mutant: 03 | 778–779; 788–789: cleavage predicted | 778–779; 788–789: cleavage not predicted |
| ITGB7 | R760H | Wild-type: 06 Mutant: 05 | 769–770: cleavage predicted | 769–770: cleavage not predicted |
| ITGB3 | D749N | Wild-type: 12 Mutant: 12 | No variation predicted | No variation predicted |
| R786Q | Wild-type: 12 Mutant: 10 | 772–773; 779–780: cleavage predicted | 772–773; 779–780: cleavage not predicted | |
| Parameters | Wild-Type ITGB1 | Mutant ITGB1 (T777M) |
|---|---|---|
| DeepCalp cleavage sites | 5 | 3 |
| Docking score | −222.61 | −206.58 |
| Confidence score | 0.8103 | 0.7561 |
| RMSD | 1.227Å | |
| H-bonds | 8 | 4 |
| Hydrophobic interactions | 6 | 7 |
| Electrostatic interactions | 9 | 2 |
| Average radius of gyrtaion | 4.72 nm | 4.19 nm |
| Parameters | Wild-Type ITGB7 | Mutant ITGB7 (R760H) |
|---|---|---|
| DeepCalp cleavage sites | 6 | 5 |
| Docking score | −222.31 | −222.50 |
| Confidence score | 0.8094 | 0.8100 |
| RMSD | 1.081Å | |
| H-bonds | 10 | 6 |
| Hydrophobic interactions | 9 | 3 |
| Electrostatic interactions | 6 | 5 |
| Average radius of gyrtaion | 3.72 nm | 4.60 nm |
| Parameters | Wild-Type ITGB3 | Mutant ITGB3 D749N | Mutant ITGB3 R786Q |
|---|---|---|---|
| DeepCalp cleavage sites | 12 | 12 | 10 |
| Docking score | −283.58 | −234.95 | −224.14 |
| Confidence score | 0.935 | 0.845 | 0.815 |
| RMSD | 1.013 | 1.071 | |
| H-bonds | 12 | 8 | 6 |
| Hydrophobic interactions | 9 | 6 | 8 |
| Electrostatic interactions | 5 | 3 | 1 |
| Average radius of gyrtaion | 4.38 nm | 4.07 nm | 4.56 nm |
| (a) | (b) | (c) | ||||
|---|---|---|---|---|---|---|
| Wild-Type | Mutant T777M | Wild-Type | Mutant R760H | Wild-Type | Mutant D749N | Mutant R786Q |
| LEU753 | - | ARG747 | - | LYS742 | LYS742 | - |
| LEU754 | - | SER749 | SER749 | LEU743 | - | - |
| MET755 | - | VAL750 | VAL750 | LEU744 | - | - |
| ILE756 | - | GLU751 | GLU751 | - | ILE745 | - |
| ILE757 | - | TYR753 | TYR753 | THR746 | THR746 | - |
| HIS758 | HIS758 | ASP754 | - | ILE747 | - | - |
| ASP759 | - | - | ARG756 | - | HIS748 | - |
| ARG761 | ARG761 | GLU757 | GLU757 | ASP749 | ASN749 | - |
| GLU762 | GLU762 | - | HIS760 | ARG750 | - | - |
| ALA764 | - | GLU764 | - | LYS751 | - | - |
| LYS765 | LYS765 | GLN767 | - | - | GLU752 | - |
| LYS768 | LYS768 | LEU768 | - | PHE753 | PHE753 | PHE753 |
| GLU769 | GLU769 | - | TRP770 | - | PHE756 | PHE756 |
| LYS770 | - | LYS771 | LYS771 | GLU757 | - | GLU757 |
| MET771 | - | - | ASN775 | GLU758 | - | GLU758 |
| ASN772 | ASN772 | - | TYR778 | ARG760 | - | ARG760 |
| ALA773 | - | LYS779 | LYS779 | ALA761 | - | ALA761 |
| LYS774 | LYS774 | SER780 | SER780 | ARG762 | - | - |
| TRP775 | TRP775 | ALA781 | ALA781 | ALA763 | - | - |
| ASP776 | ASP776 | - | - | - | - | LYS764 |
| THR777 | MET777 | - | - | - | - | TRP765 |
| - | GLY778 | THR767 | - | THR767 | ||
| GLU779 | GLU779 | ALA768 | - | ALA768 | ||
| ASN780 | ASN780 | ASN769 | - | - | ||
| - | PRO781 | ASN770 | - | - | ||
| TYYR783 | - | PRO771 | - | - | ||
| - | LYS784 | LEU772 | - | - | ||
| - | ALA786 | - | - | TYR773 | ||
| - | VAL787 | LYS774 | - | LYS774 | ||
| - | THR788 | GLU775 | - | - | ||
| - | THR789 | ALA776 | - | - | ||
| THR777 | THR777 | THR777 | ||||
| SER778 | SER778 | SER778 | ||||
| - | - | THR779 | ||||
| - | PHE780 | PHE780 | ||||
| - | THR781 | THR781 | ||||
| - | ASN782 | ASN782 | ||||
| - | ILE783 | - | ||||
| - | THR784 | - | ||||
| TYR785 | TYR785 | TYR785 | ||||
| - | ARG786 | GLN786 | ||||
| - | GLY787 | GLY787 | ||||
| THR788 | THR788 | THR788 | ||||
| Sl. No | Protein Substrate | Selected Mutation | Calpain Cleavage Sites | Calpain Type |
|---|---|---|---|---|
| 1. | ITGB1 | T777M | 771 (MN), 777 (TG), 778 (GE), 789 (TV), 791 (VN) | Calpain-2 (m-calpain) |
| 2. | ITGB7 | R760H | 746 (YR), 760 (RF), 765 (QQ), 766 (QQ), 769 (NW), 770 (WK), 773 (DS), 774 (SN), 778 (YK), 784 (TT), 785 (TI) | Calpain-2 (m-calpain) |
| 3. | ITGB3 | D749N R786Q | 761 (AR), 767 (TA), 773 (YK), 780 (FT), 785 (YR) | Calpain-1 (µ-calpain) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizhakethil, R.V.; Varma, A.K.; Barage, S.H.; Ramesh Kumar, N.; Nagarajan, K.; Santhosh Kumar, A.W.; Kamble, S.S. Repercussions of the Calpain Cleavage-Related Missense Mutations in the Cytosolic Domains of Human Integrin-β Subunits on the Calpain–Integrin Signaling Axis. Int. J. Mol. Sci. 2025, 26, 4246. https://doi.org/10.3390/ijms26094246
Kizhakethil RV, Varma AK, Barage SH, Ramesh Kumar N, Nagarajan K, Santhosh Kumar AW, Kamble SS. Repercussions of the Calpain Cleavage-Related Missense Mutations in the Cytosolic Domains of Human Integrin-β Subunits on the Calpain–Integrin Signaling Axis. International Journal of Molecular Sciences. 2025; 26(9):4246. https://doi.org/10.3390/ijms26094246
Chicago/Turabian StyleKizhakethil, Reshma V., Ashok K. Varma, Sagar H. Barage, Neelmegam Ramesh Kumar, Kayalvizhi Nagarajan, Aruni Wilson Santhosh Kumar, and Shashank S. Kamble. 2025. "Repercussions of the Calpain Cleavage-Related Missense Mutations in the Cytosolic Domains of Human Integrin-β Subunits on the Calpain–Integrin Signaling Axis" International Journal of Molecular Sciences 26, no. 9: 4246. https://doi.org/10.3390/ijms26094246
APA StyleKizhakethil, R. V., Varma, A. K., Barage, S. H., Ramesh Kumar, N., Nagarajan, K., Santhosh Kumar, A. W., & Kamble, S. S. (2025). Repercussions of the Calpain Cleavage-Related Missense Mutations in the Cytosolic Domains of Human Integrin-β Subunits on the Calpain–Integrin Signaling Axis. International Journal of Molecular Sciences, 26(9), 4246. https://doi.org/10.3390/ijms26094246