
Pike: OTU-Level Analysis for Oxford Nanopore Amplicon Metagenomics

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Raw Data Processing
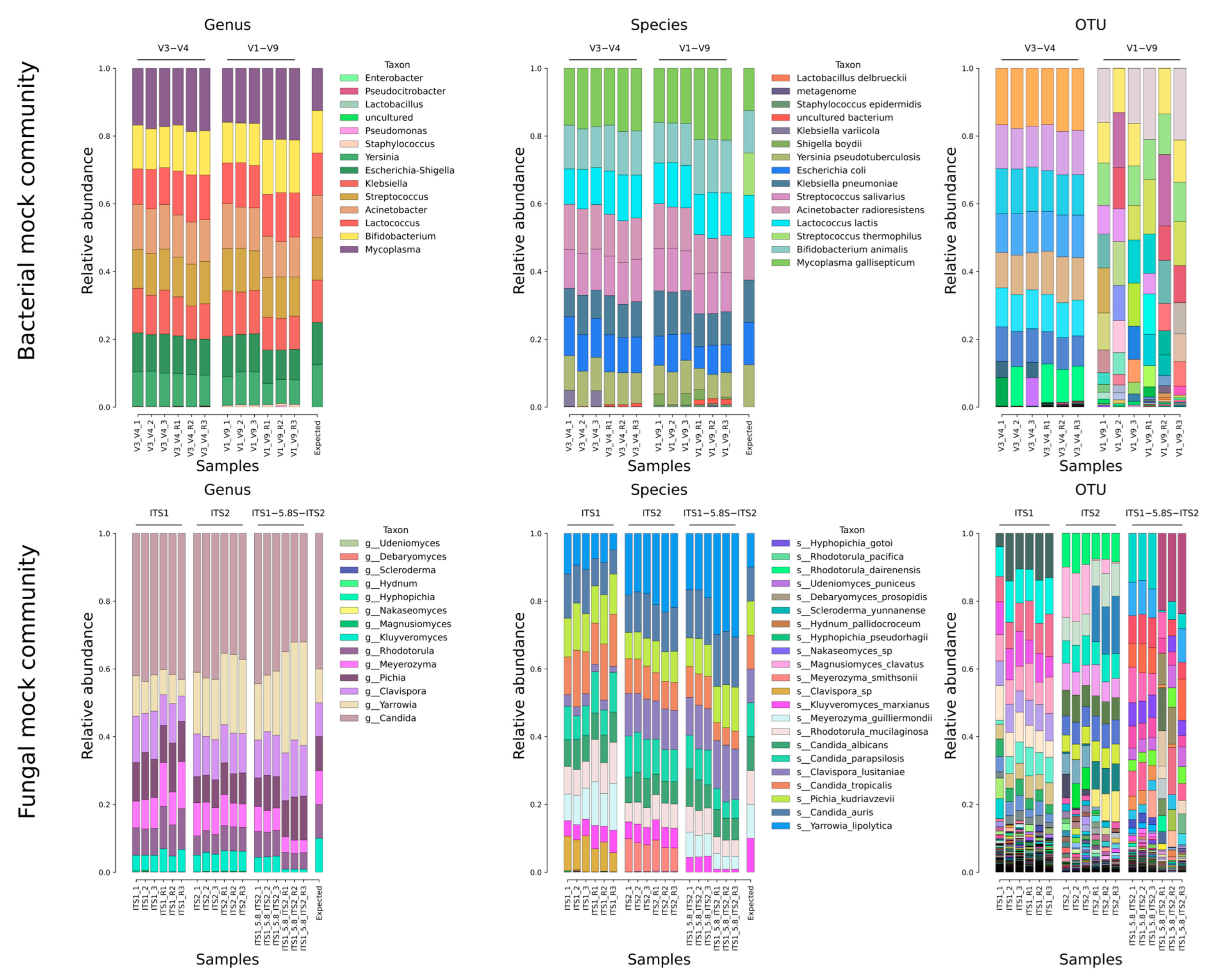
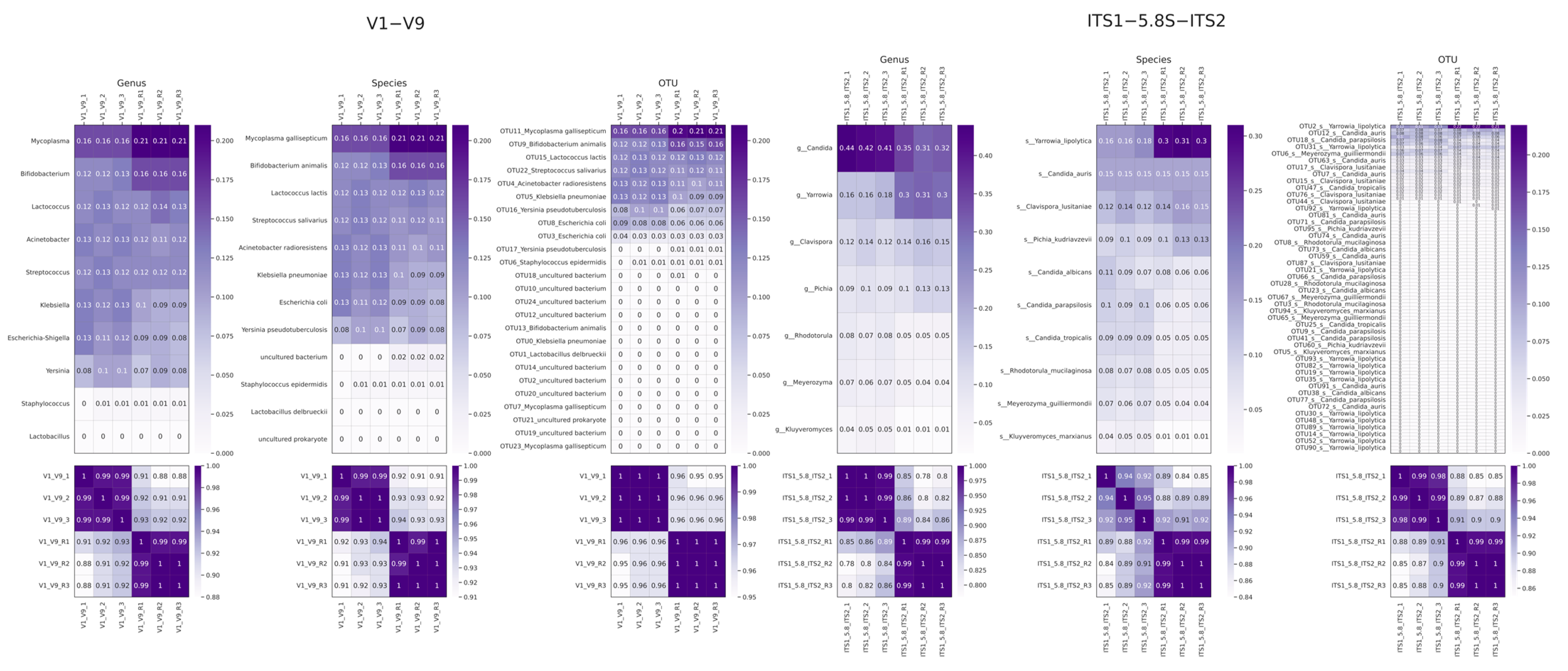
2.2. Mock Community Analysis
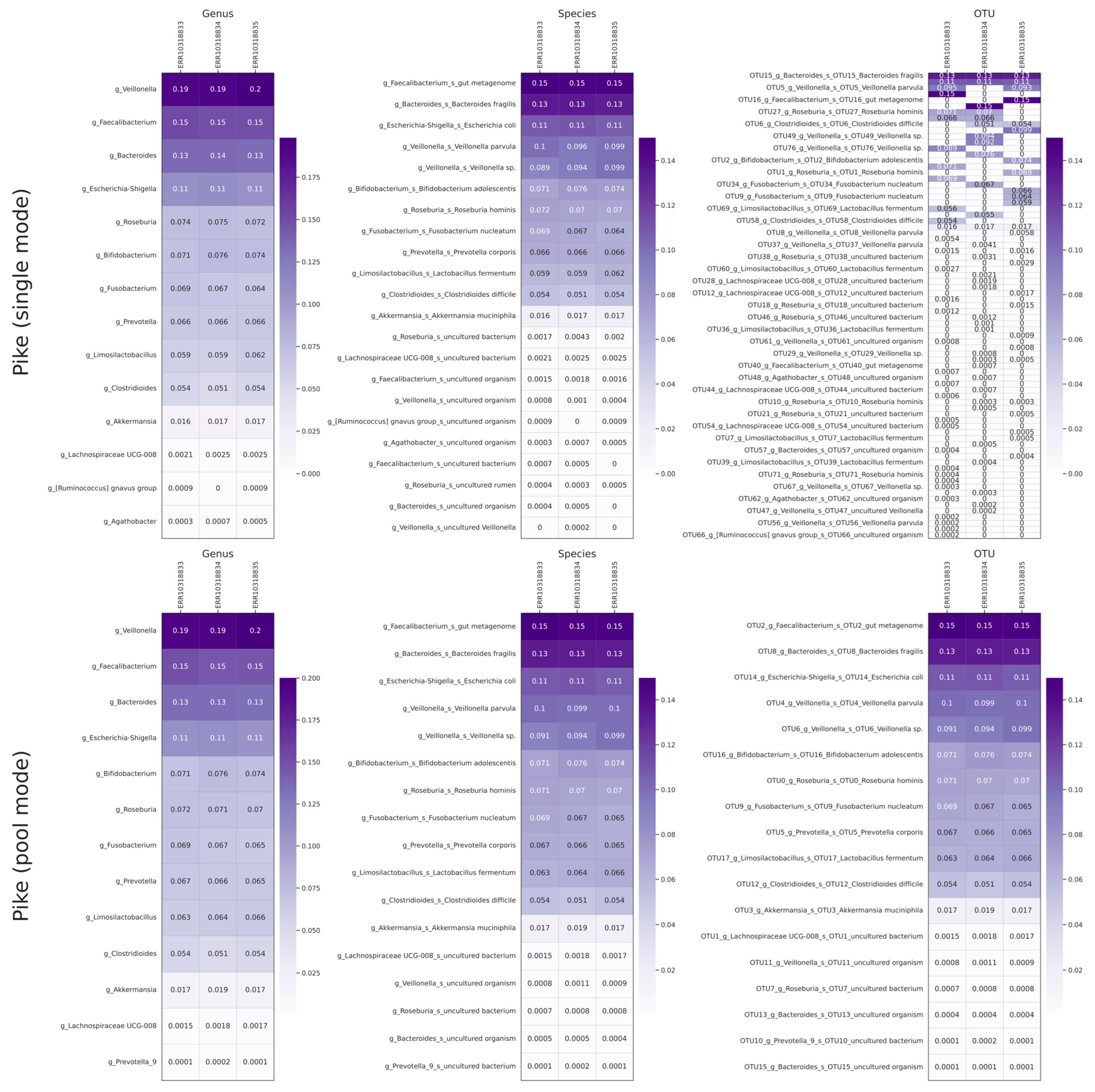
2.3. Analysis of External Data
2.4. Benchmarking and Comparison with Alternative Tools
2.5. Discussion Summary
3. Materials and Methods
3.1. Experiment Design
3.1.1. Amplification of 16S rRNA
3.1.2. Amplification of Fungal ITS1, ITS2 Region
3.2. Amplicon Compositions and Reamplification
Library Preparation and Sequencing
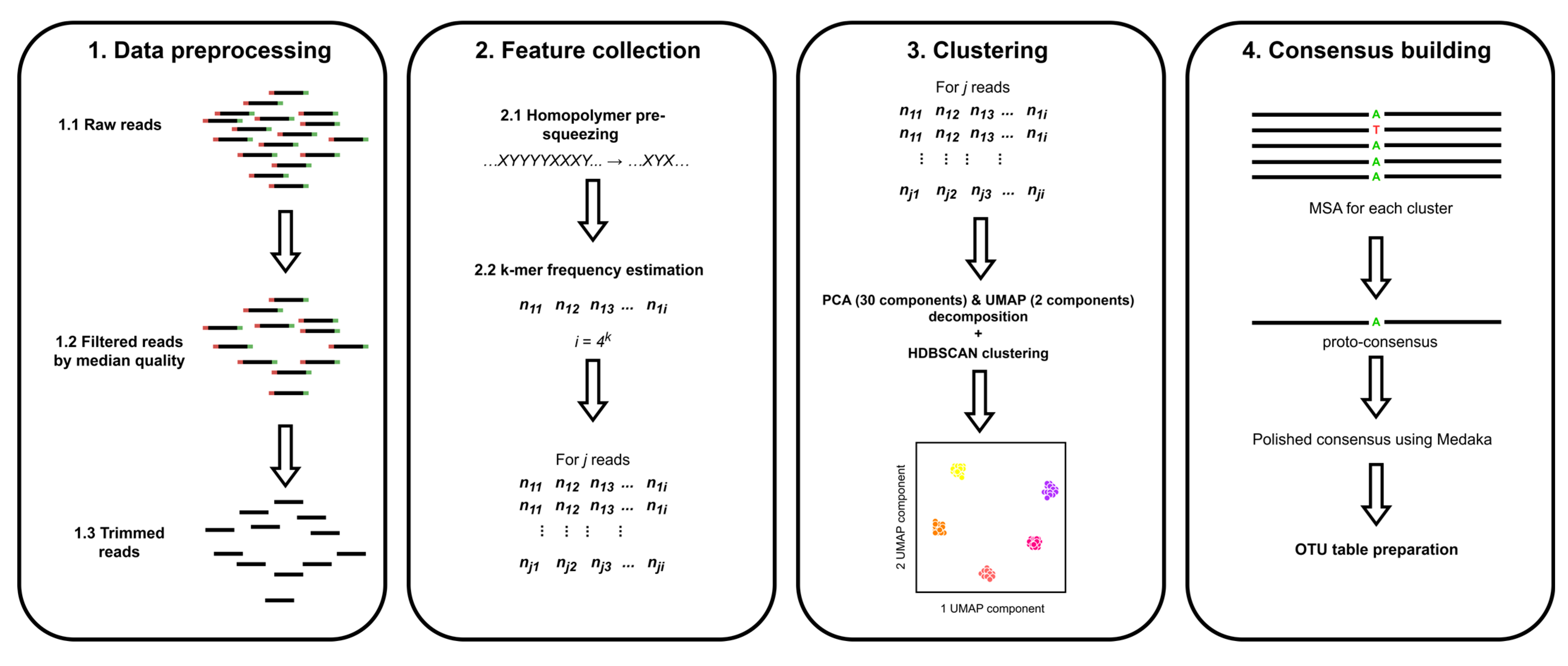
3.3. Algorithm Explanation
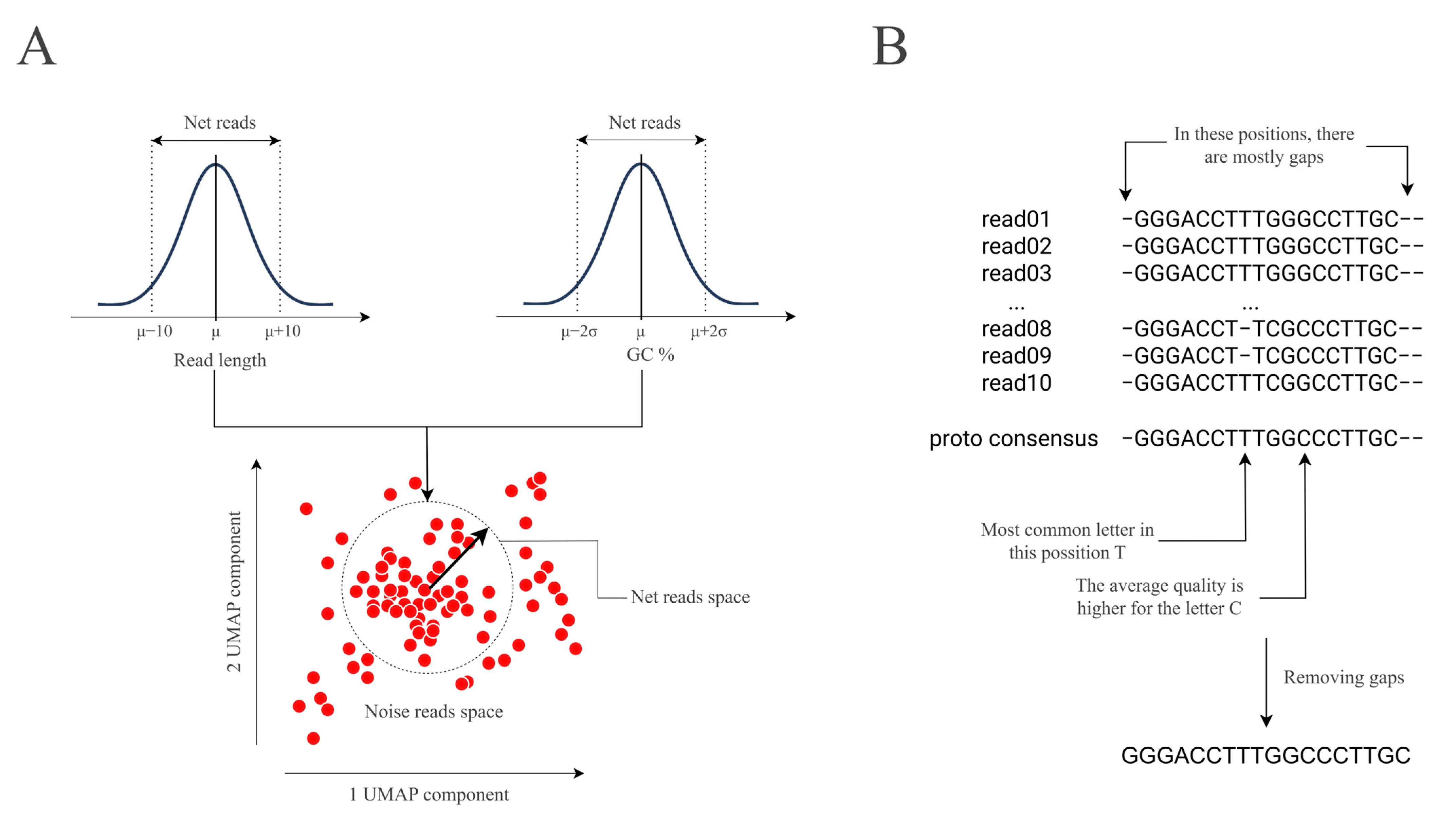
3.3.1. Reads Preparation
3.3.2. Feature Collection
3.3.3. Clustering
3.3.4. Consensus Building
3.3.5. Mode Explanation
3.3.6. Taxonomy Identification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Heikema, A.P.; Horst-Kreft, D.; Boers, S.A.; Jansen, R.; Hiltemann, S.D.; de Koning, W.; Kraaij, R.; de Ridder, M.A.J.; van Houten, C.B.; Bont, L.J.; et al. Comparison of illumina versus nanopore 16S rRNA gene sequencing of the human nasal microbiota. Genes 2020, 11, 1105. [Google Scholar] [CrossRef] [PubMed]
- Loit, K.; Adamson, K.; Bahram, M.; Puusepp, R.; Anslan, S.; Kiiker, R.; Drenkhan, R.; Tedersoo, L. Relative performance of MinION (Oxford Nanopore Technologies) versus Sequel (Pacific Biosciences) third-generation sequencing instruments in identification of agricultural and forest fungal pathogens. Appl. Environ. Microbiol. 2019, 85, e01368-19. [Google Scholar] [CrossRef]
- Stevens, B.M.; Creed, T.B.; Reardon, C.L.; Manter, D.K. Comparison of Oxford Nanopore Technologies and Illumina MiSeq sequencing with mock communities and agricultural soil. Sci. Rep. 2023, 13, 9323. [Google Scholar] [CrossRef]
- Sawaswong, V.; Chanchaem, P.; Klomkliew, P.; Rotcheewaphan, S.; Meesawat, S.; Kemthong, T.; Kaewparuehaschai, M.; Noradechanon, K.; Ekatat, M.; Kanitpun, R.; et al. Full-length 16S rDNA sequencing based on Oxford Nanopore Technologies revealed the association between gut-pharyngeal microbiota and tuberculosis in cynomolgus macaques. Sci. Rep. 2024, 14, 3404. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Ma, S.; Cao, J.; Liao, H.; Huang, Q.; Chen, W. The newest Oxford Nanopore R10.4.1 full-length 16S rRNA sequencing enables the accurate resolution of species-level microbial community profiling. Appl. Environ. Microbiol. 2023, 89, e00605-23. [Google Scholar] [CrossRef]
- Langsiri, N.; Worasilchai, N.; Irinyi, L.; Jenjaroenpun, P.; Wongsurawat, T.; Luangsa-Ard, J.J.; Meyer, W.; Chindamporn, A. Targeted sequencing analysis pipeline for species identification of human pathogenic fungi using long-read nanopore sequencing. IMA Fungus 2023, 14, 18. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A species-level analysis of 16S rRNA nanopore sequencing data. Bioinformatics 2021, 37, 1600–1601. [Google Scholar] [CrossRef] [PubMed]
- Schacksen, P.S.; Østergaard, S.K.; Eskildsen, M.H.; Nielsen, J.L. Complete pipeline for Oxford Nanopore Technology amplicon sequencing (ONT-AmpSeq): From pre-processing to creating an operational taxonomic unit table. FEBS Open Bio 2024, 14, 1779–1787. [Google Scholar] [CrossRef]
- Ohta, A.; Nishi, K.; Hirota, K.; Matsuo, Y. Using nanopore sequencing to identify fungi from clinical samples with high phylogenetic resolution. Sci. Rep. 2023, 13, 9785. [Google Scholar] [CrossRef] [PubMed]
- Nanoporetech. Nanoporetech. Nanoporetech/Medaka: Sequence Correction Provided by ONT Research; GitHub: San Francisco, CA, USA. In Nanoporetech/Medaka: Sequence Correction Provided by ONT Research; GitHub: San Francisco, CA, USA; Available online: https://github.com/nanoporetech/medaka (accessed on 13 July 2023).
- epi2me-labs. Epi2me-Labs/WF-Metagenomics: Metagenomic Classification of Long-Read Sequencing Data; GitHub: San Francisco, CA, USA; Available online: https://github.com/epi2me-labs/wf-metagenomics (accessed on 12 July 2024).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Garrido-Sanz, L.; Senar, M.À.; Piñol, J. Drastic reduction of false positive species in samples of insects by intersecting the default output of two popular metagenomic classifiers. PLoS ONE 2022, 17, e0275790. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 July 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Francisco, E.C.; Ribeiro, F.d.C.; Junior, J.N.A.; Pedoni, D.B.; da Matta, D.A.; Dolande, M.; Melo, A.S.d.A.; Lima, R.F.; Aquino, V.R.; Corzo-León, D.E.; et al. Emergence of cryptic species and clades of Meyerozyma guilliermondii species complex exhibiting limited in vitro susceptibility to antifungals in patients with candidemia. Microbiol. Spectr. 2023, 11, e05115-22. [Google Scholar] [CrossRef]
- Nicholls, S.M.; Quick, J.C.; Tang, S.; Loman, N.J. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience 2019, 8, giz043. [Google Scholar] [CrossRef] [PubMed]
- Gand, M.; Bloemen, B.; Vanneste, K.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Comparison of 6 DNA extraction methods for isolation of high yield of high molecular weight DNA suitable for shotgun metagenomics Nanopore sequencing to detect bacteria. BMC Genom. 2023, 24, 438. [Google Scholar] [CrossRef]
- Cuscó, A.; Catozzi, C.; Viñes, J.; Sanchez, A.; Francino, O. Microbiota profiling with long amplicons using Nanopore sequencing: Full-length 16S rRNA gene and the 16S-ITS-23S of the rrn operon. F1000Research 2019, 7, 1755. [Google Scholar] [CrossRef]
- Szoboszlay, M.; Schramm, L.; Pinzauti, D.; Scerri, J.; Sandionigi, A.; Biazzo, M. Nanopore is preferable over illumina for 16S amplicon sequencing of the gut microbiota when species-level taxonomic classification, accurate estimation of richness, or focus on rare taxa is required. Microorganisms 2023, 11, 804. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, pipelines, parameters: Issues in 16S rRNA gene sequencing. mSphere 2021, 6, 10-1128. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA Genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; Volume 18, pp. 315–322. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Chiou, C.-S.; Chen, B.-H.; Wang, Y.-W.; Kuo, N.-T.; Chang, C.-H.; Huang, Y.-T. Correcting modification-mediated errors in nanopore sequencing by nucleotide demodification and reference-based correction. Commun. Biol. 2023, 6, 1215. [Google Scholar] [CrossRef] [PubMed]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, K.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef]
- Rolling, T.; Zhai, B.; Frame, J.; Hohl, T.M.; Taur, Y. Customization of a DADA2-based pipeline for fungal internal transcribed spacer 1 (ITS1) amplicon data sets. JCI Insight 2021, 7, 151663. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: A novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Rodrigues, J.F.M.; Schmidt, T.S.B.; Tackmann, J.; von Mering, C. MAPseq: Highly efficient k-mer search with confidence estimates, for rRNA sequence analysis. Bioinformatics 2017, 33, 3808–3810. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Allard, G.; Ryan, F.J.; Jeffery, I.B.; Claesson, M.J. SPINGO: A rapid species-classifier for microbial amplicon sequences. BMC Bioinform. 2015, 16, 324. [Google Scholar] [CrossRef]
- Krehenwinkel, H.; Wolf, M.; Lim, J.Y.; Rominger, A.J.; Simison, W.B.; Gillespie, R.G. Estimating and mitigating amplification bias in qualitative and quantitative arthropod metabarcoding. Sci. Rep. 2017, 7, 17668. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Single Mode | Pool Mode | ||||||
|---|---|---|---|---|---|---|---|
| Species | Expected | ERR10318833 | ERR10318834 | ERR10318835 | ERR10318833 | ERR10318834 | ERR10318835 |
| Veillonella rogosae | 0.1587 | 0.190446 | 0.191516 | 0.1983 | 0.1927 | 0.194628 | 0.200128 |
| Faecalibacterium prausnitzii | 0.1763 | 0.152488 | 0.152488 | 0.1489 | 0.1498 | 0.1459 | 0.1466 |
| Bacteroides fragilis | 0.0994 | 0.1348 | 0.1350 | 0.1328 | 0.1344 | 0.1343 | 0.1328 |
| Escherichia coli | 0.1212 | 0.1090 | 0.1101 | 0.1066 | 0.1088 | 0.1098 | 0.1069 |
| Bifidobacterium adolescentis | 0.0878 | 0.0707 | 0.0760 | 0.0745 | 0.0708 | 0.0762 | 0.0742 |
| Roseburia hominis | 0.0989 | 0.0743 | 0.0749 | 0.0721 | 0.0718 | 0.0708 | 0.0703 |
| Fusobacterium nucleatum | 0.0749 | 0.0693 | 0.0669 | 0.0645 | 0.0692 | 0.0668 | 0.0646 |
| Prevotella corporis | 0.0498 | 0.0663 | 0.0658 | 0.0661 | 0.0667 | 0.0655 | 0.0654 |
| Lactobacillus fermentum | 0.0963 | 0.0590 | 0.0587 | 0.0620 | 0.0629 | 0.0643 | 0.0661 |
| Clostridioides difficile | 0.0262 | 0.0540 | 0.0510 | 0.0536 | 0.0543 | 0.0511 | 0.0539 |
| Akkermansia muciniphila | 0.0970 | 0.0164 | 0.0174 | 0.0167 | 0.0168 | 0.0187 | 0.0172 |
| Salmonella enterica | 0.00009 | 0 | 0 | 0 | 0 | 0 | 0 |
| Clostridium perfringens | 0.000002 | 0 | 0 | 0 | 0 | 0 | 0 |
| Enterococcus faecalis | 0.000009 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fungal Composition | Bacterial Composition |
|---|---|
| Clavispora lusitaniae Candida tropicalis Kluyveromyces marxianus Candida auris Pichia kudryavtseva Candida albicans Rhodotorula mucilaginosa Candida parapsilosis Meyerozyma guilliermondii Yarrowia lipolytica | Streptococcus thermophilus Lactococcus lactis Bifidobacterium animalis Escherichia coli Yersinia pseudotuberculosis Klebsiella pneumoniae Acinetobacter baumannii Mycoplasma gallisepticum |
| Mock Community Type | Amplicon Region | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) | Source |
|---|---|---|---|---|
| Bacteria | V3–V4 | CCTACGGGNGGCWGCAG | GACTACHVGGGTATCTAATCC | [28] |
| V1–V9 | AGRGTTYGATYMTGGCTCAG | CGGYTACCTTGTTACGACTT | [29] | |
| Fungi | ITS1 | GGAAGTAAAAGTCGTAACAAGG | GCTGCGTTCTTCATCGATGC | |
| ITS2 | GCATCGATGAAGAACGCAGC | TCCTCCGCTTATTGATATGC | [30] | |
| ITS1-5.8S-ITS2 | GGAAGTAAAAGTCGTAACAAGG | TCCTCCGCTTATTGATATGC |
| Bacteria | Fungi | ||||
|---|---|---|---|---|---|
| 16S V3–V4 | 16S V1–V9 | ITS1 | ITS2 | ITS1-5.8S-ITS2 | |
| Minimum expected length (bp) | 350 | 900 | 100 | 100 | 300 |
| Maximum expected length (bp) | 600 | 1600 | 500 | 500 | 1200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivonos, D.V.; Fedorov, D.E.; Konanov, D.N.; Vvedensky, A.V.; Sonets, I.V.; Korneenko, E.V.; Speranskaya, A.S.; Ilina, E.N. Pike: OTU-Level Analysis for Oxford Nanopore Amplicon Metagenomics. Int. J. Mol. Sci. 2025, 26, 4168. https://doi.org/10.3390/ijms26094168
Krivonos DV, Fedorov DE, Konanov DN, Vvedensky AV, Sonets IV, Korneenko EV, Speranskaya AS, Ilina EN. Pike: OTU-Level Analysis for Oxford Nanopore Amplicon Metagenomics. International Journal of Molecular Sciences. 2025; 26(9):4168. https://doi.org/10.3390/ijms26094168
Chicago/Turabian StyleKrivonos, Danil V., Dmitry E. Fedorov, Dmitry N. Konanov, Andrey V. Vvedensky, Ignat V. Sonets, Elena V. Korneenko, Anna S. Speranskaya, and Elena N. Ilina. 2025. "Pike: OTU-Level Analysis for Oxford Nanopore Amplicon Metagenomics" International Journal of Molecular Sciences 26, no. 9: 4168. https://doi.org/10.3390/ijms26094168
APA StyleKrivonos, D. V., Fedorov, D. E., Konanov, D. N., Vvedensky, A. V., Sonets, I. V., Korneenko, E. V., Speranskaya, A. S., & Ilina, E. N. (2025). Pike: OTU-Level Analysis for Oxford Nanopore Amplicon Metagenomics. International Journal of Molecular Sciences, 26(9), 4168. https://doi.org/10.3390/ijms26094168