
GAL-201 as a Promising Amyloid-β-Targeting Small-Molecule Approach for Alzheimer’s Disease Treatment: Consistent Effects on Synaptic Plasticity, Behavior and Neuroinflammation

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
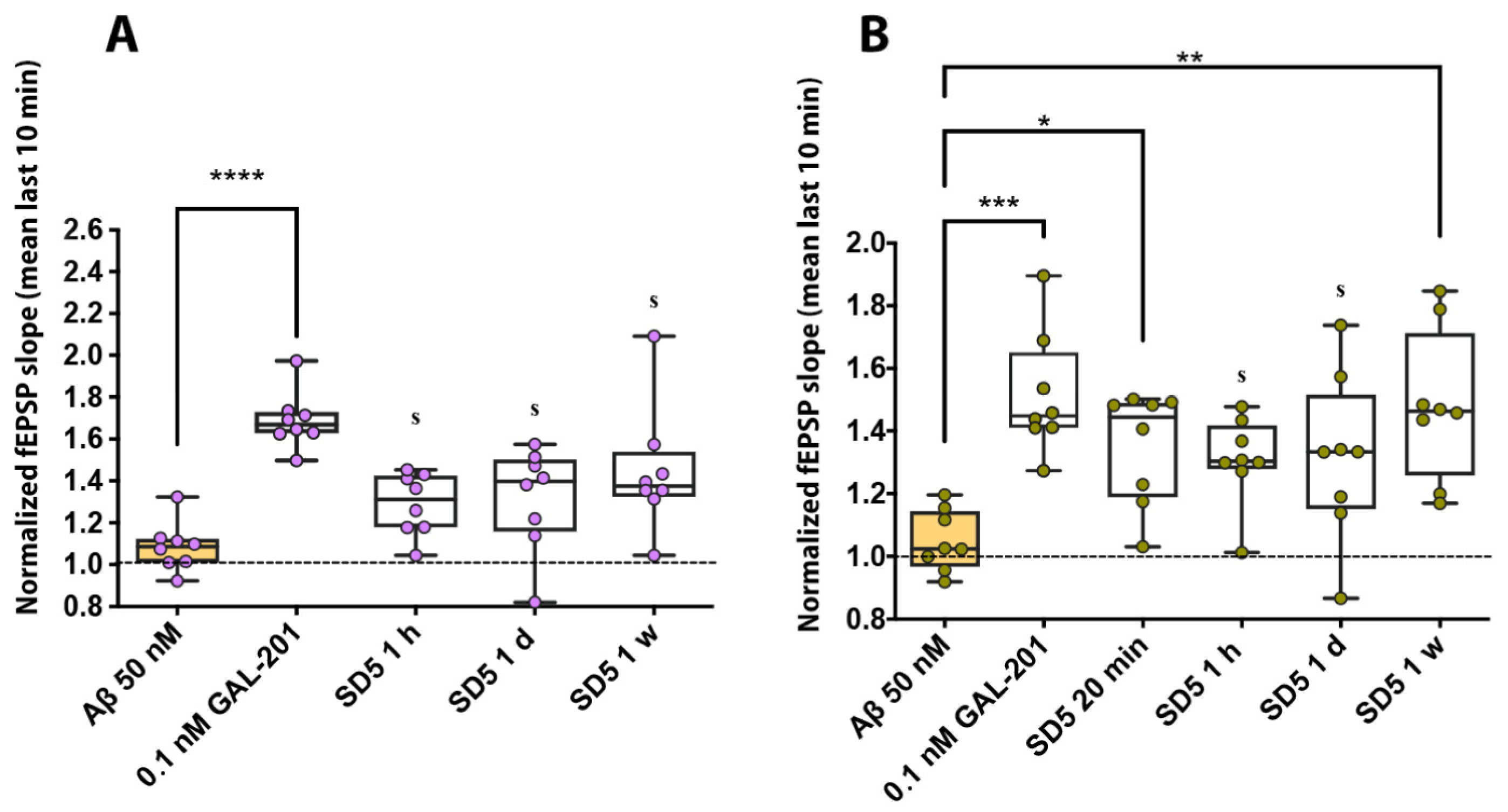
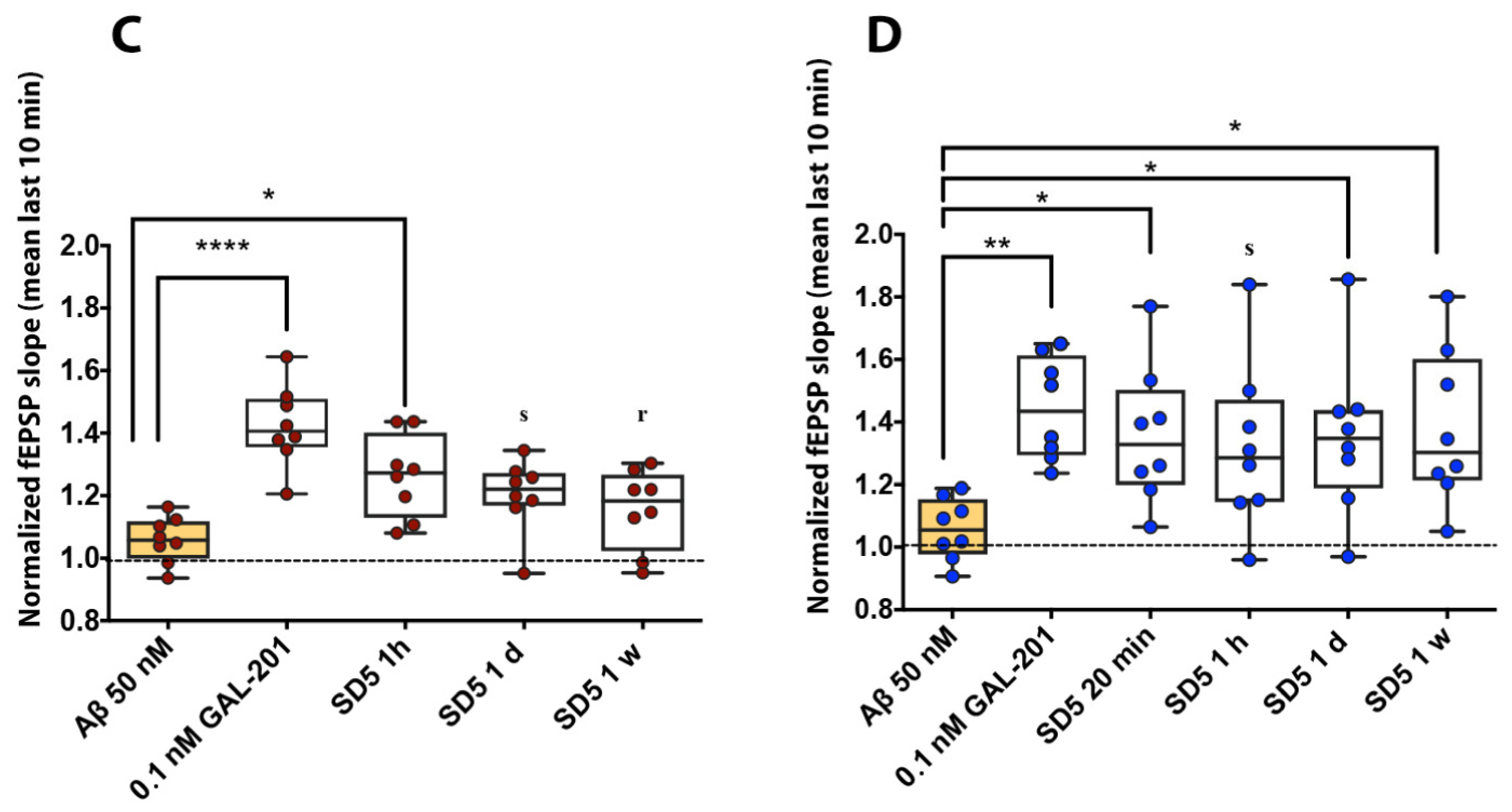
2.1. Electrophysiology in Hippocampal Brain Slices
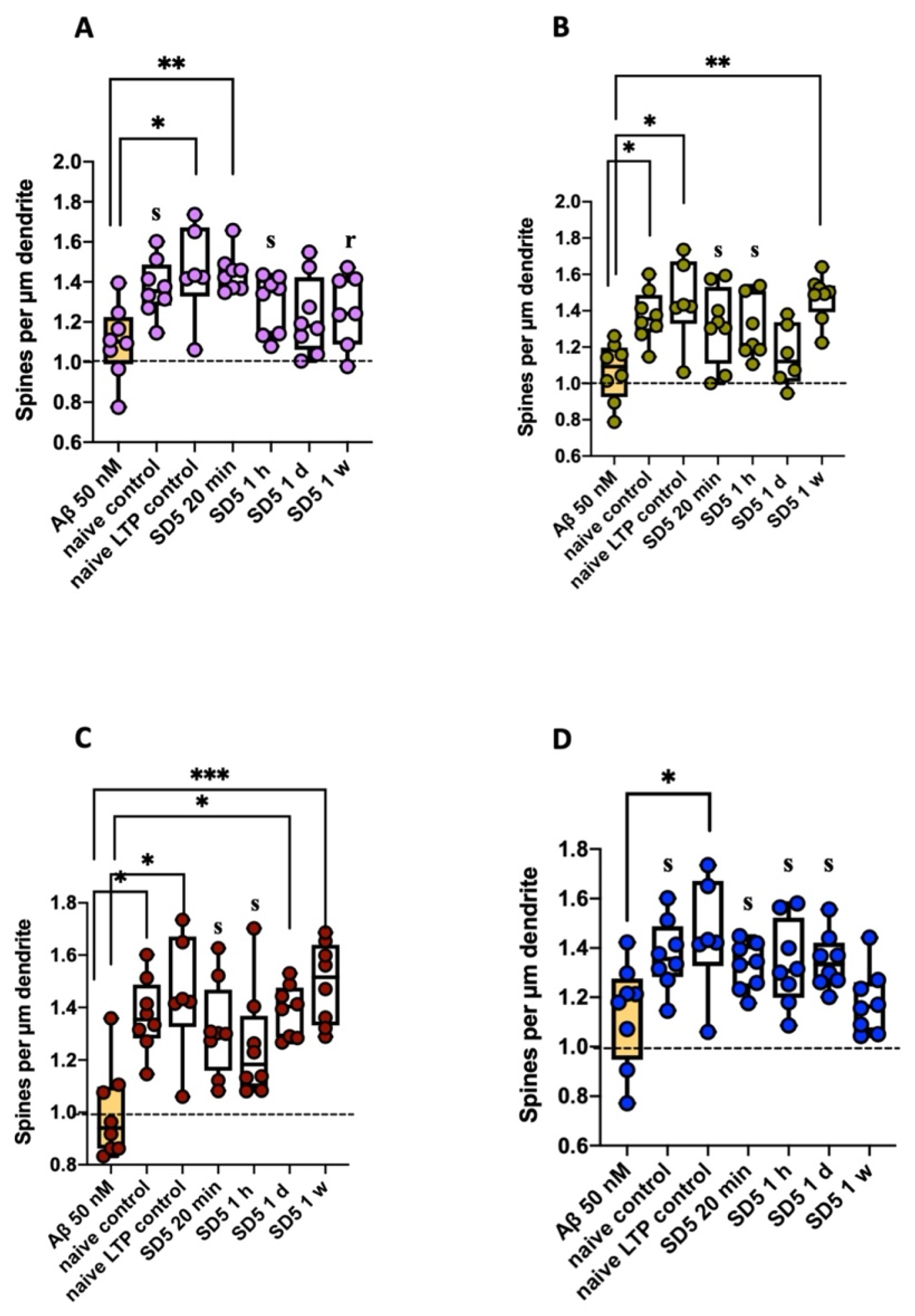
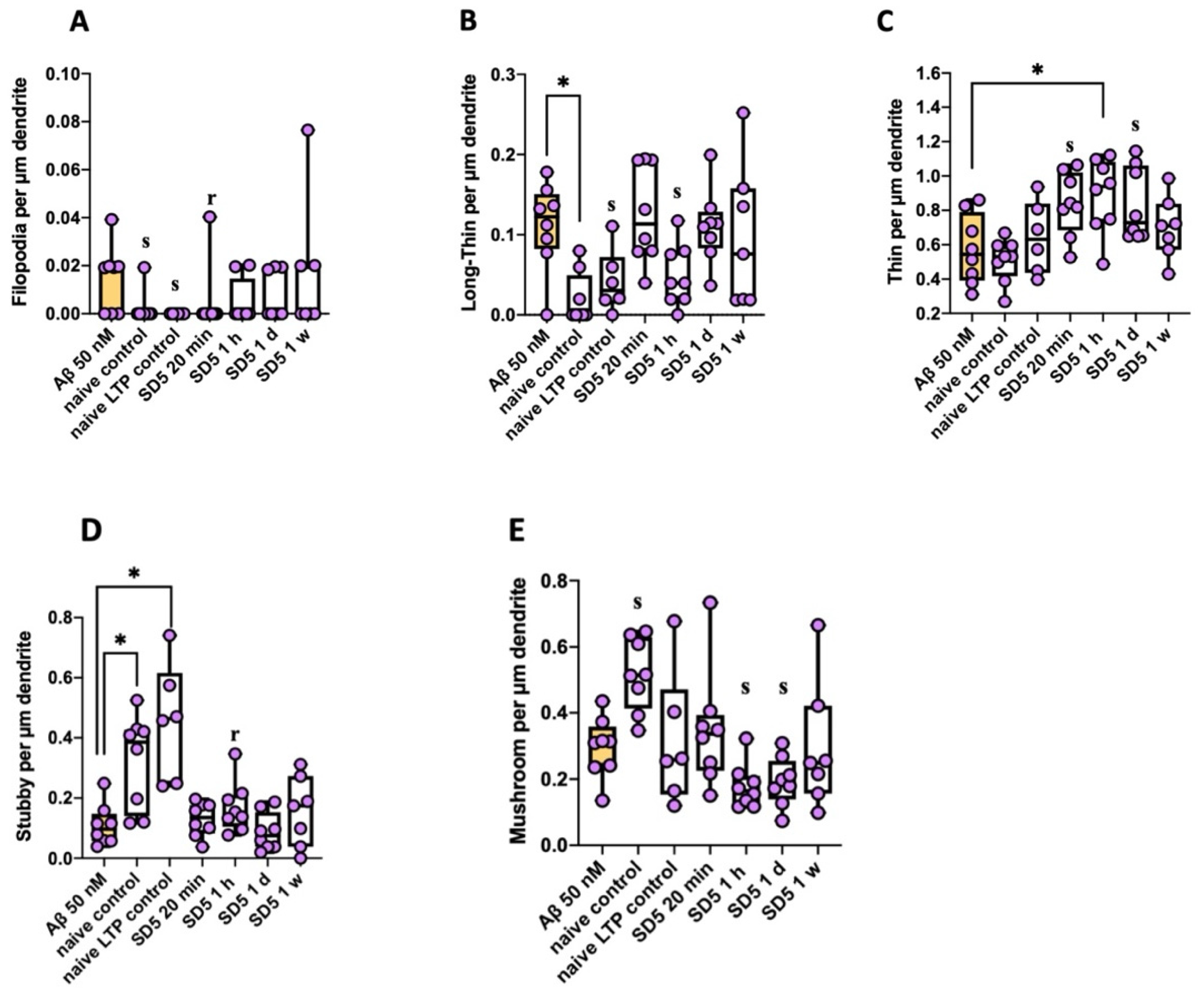
2.2. Analysis of Spine Dynamics
2.2.1. Spine Density
2.2.2. Spine Morphology (According to Risher [26])
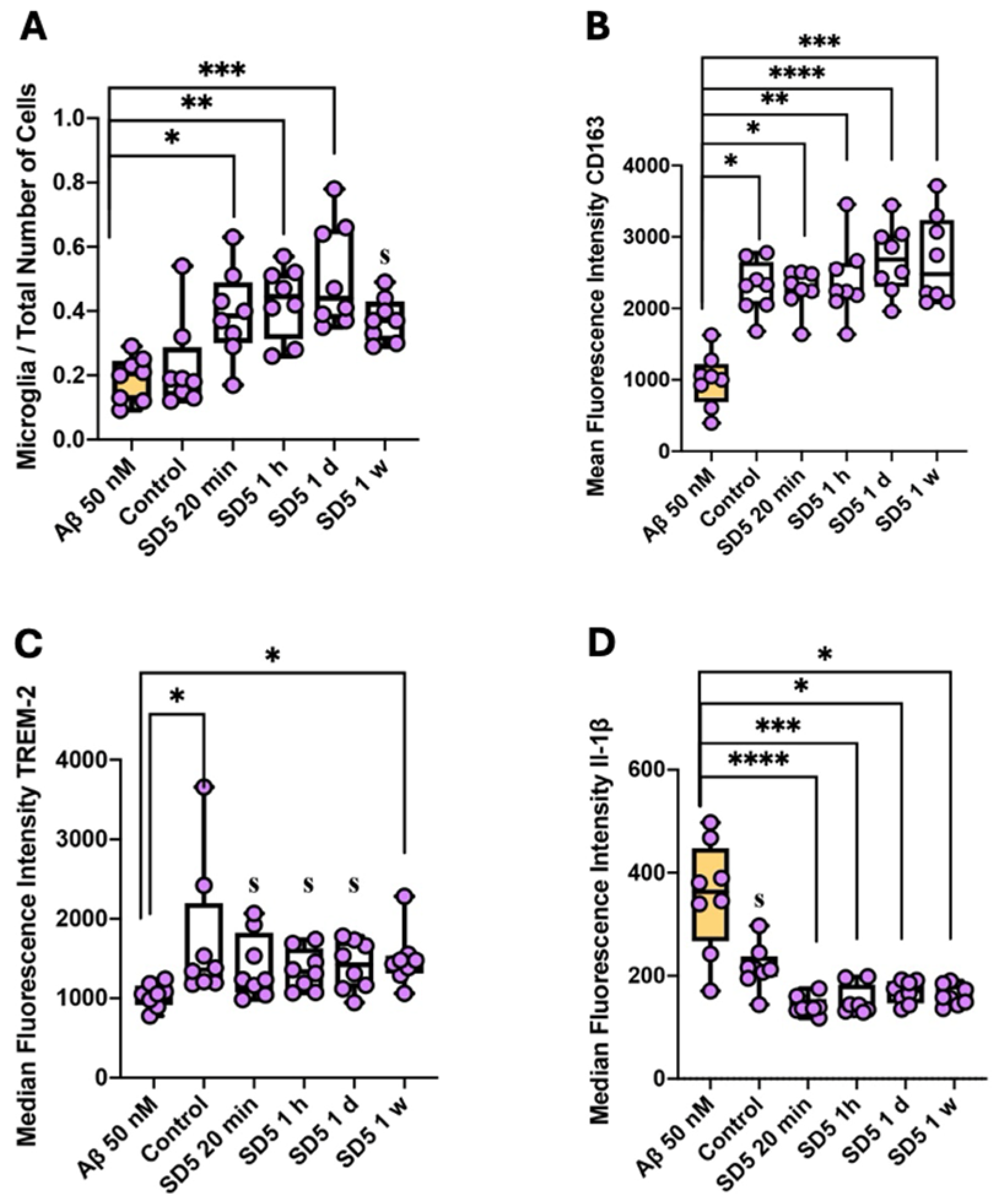
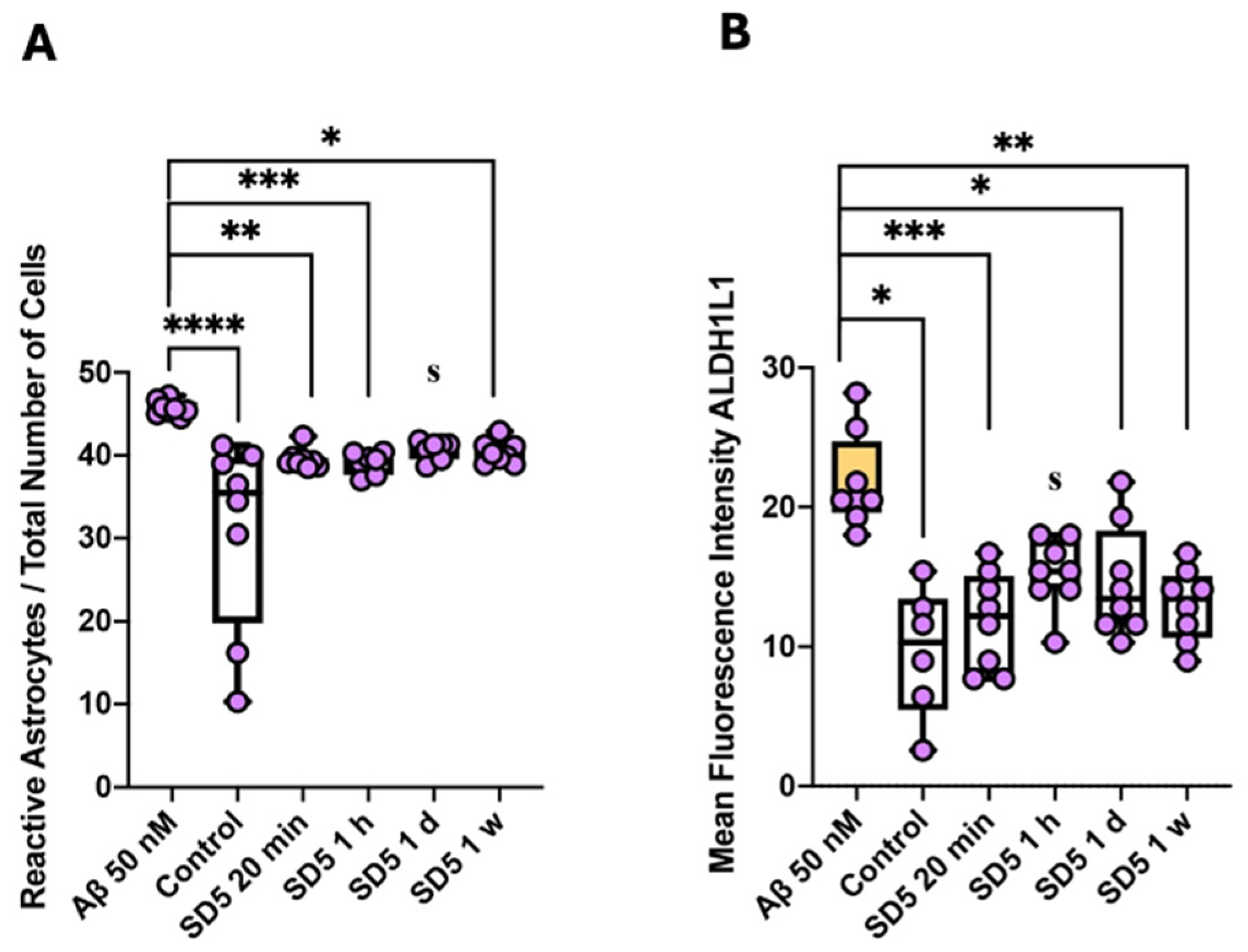
2.3. Activation of Microglia and Astrocytes
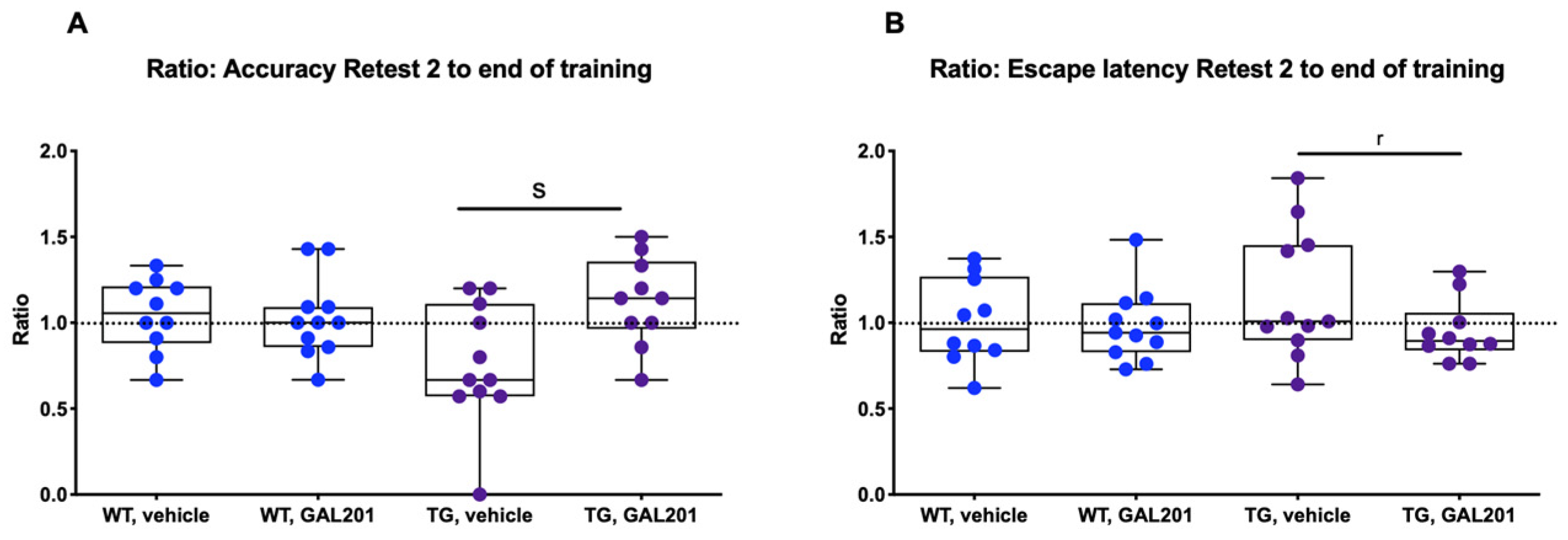
2.4. Cognitive Performance Was Improved in tgArcSwe Mice After Acute Treatment with GAL-201
2.5. Changes in the Occurrence of Plaques in tgArcSwe Mice After Acute Treatment with GAL-201
3. Discussion
3.1. Consistency of the Results
3.2. Interpretation of the Dilution Series
3.3. Influence on Spine Dynamics
3.4. Impact on Behavior
3.5. Influence on Reactive Gliosis and Neuroinflammation
3.6. Summary
4. Materials and Methods
4.1. Animals
4.2. Brain Slice Preparation
4.3. Electrophysiological Recording (Long-Term Potentiation)
4.4. Preparation of Amyloid-β, GAL-201 and Serial Dilution
4.4.1. Amyloid-β
4.4.2. GAL-201
4.4.3. Serial Dilution (GAL-201/AβSD5)
4.5. Analysis of Spine Dynamics (Golgi Staining)
4.6. Activation of Microglia and Astrocytes (Fluorescence-Activated Cell Sorting)
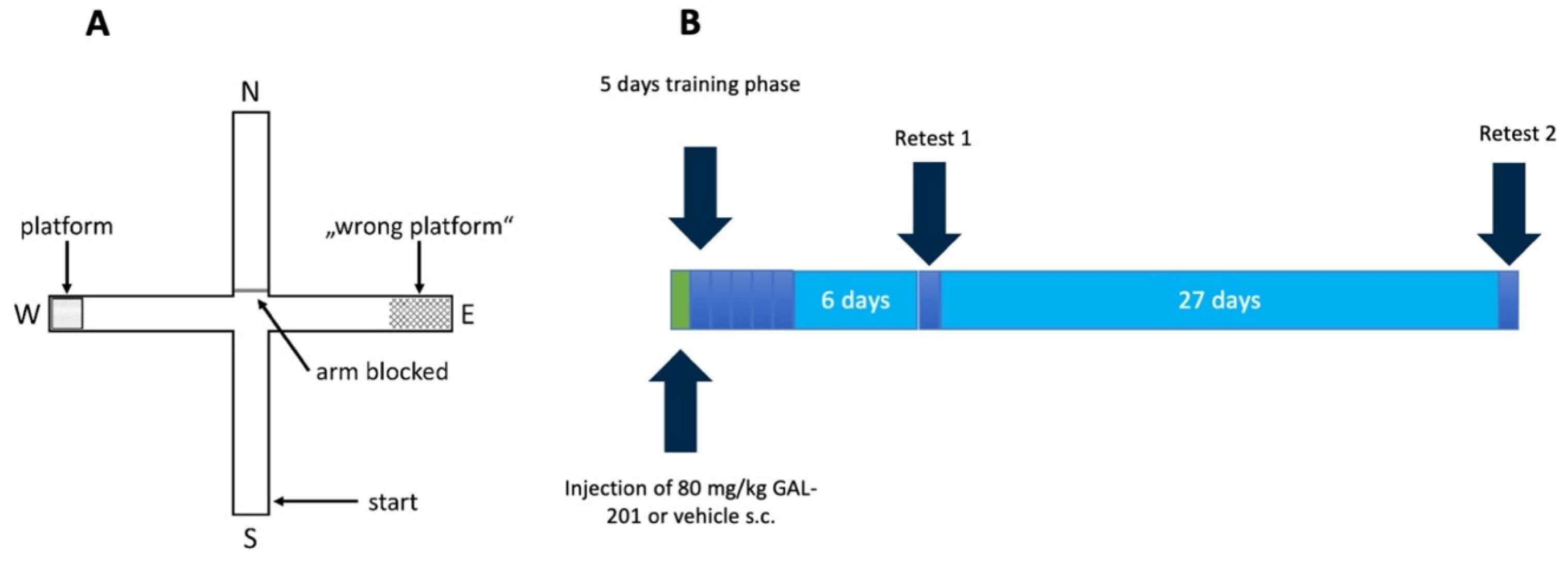
4.7. Behavioral Test
4.7.1. Preparation and Administration of GAL-201
4.7.2. Water Cross Maze
- Escape latency: time the mouse needed to reach the platform.
- Accuracy: percentage of trials per day in which the mouse found the platform directly without entering other arms.
4.8. Visualization of Plaque Formation (Methoxy-X04 Staining)
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| aCSF | artificial cerebrospinal fluid |
| AD | Alzheimer’s disease |
| ALDH1L1 | aldehyde dehydrogenase 1 family member L1 |
| AUC | area under the receiver operating characteristic |
| AUCCi | 95% confidence interval of the AUC |
| BSA | bovine serum albumin |
| CD11b | cluster of differentiation 11b |
| CD45 | cluster of differentiation 45 |
| CD163 | cluster of differentiation 163 |
| DAM | disease-associated microglia |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | dimethyl sulfoxide |
| EDTA | ethylenediaminetetraacetic acid |
| EL | escape latency |
| F4/80 | EMR1 (EGF-like module-containing-mucin-like hormone receptor-like 1) |
| FACS | fluorescence-activated cell sorting |
| fEPSP | field excitatory postsynaptic potential |
| GFAP | glial fibrillary acidic protein |
| HBSS | Hanks’ Balanced Salt Solution |
| IL-1β | interleukin-1β |
| LTP | long-term potentiation |
| P2RY12 | purinergic receptor P2Y12 |
| PBS | phosphate-buffered saline |
| PFA | paraformaldehyde |
| TG | transgenic |
| TMEM119 | transmembrane protein 119 |
| TREM-2 | triggering receptor expressed on myeloid cells 2 |
| WCM | water cross maze |
| WT | wildtype |
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- FDA. FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 27 July 2023).
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef]
- Liu, Y.H.; Bu, X.L.; Liang, C.R.; Wang, Y.R.; Zhang, T.; Jiao, S.S.; Zeng, F.; Yao, X.Q.; Zhou, H.D.; Deng, J.; et al. An N-terminal antibody promotes the transformation of amyloid fibrils into oligomers and enhances the neurotoxicity of amyloid-beta: The dust-raising effect. J. Neuroinflamm. 2015, 12, 153. [Google Scholar] [CrossRef]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Wittnam, J.L.; Portelius, E.; Zetterberg, H.; Gustavsson, M.K.; Schilling, S.; Koch, B.; Demuth, H.U.; Blennow, K.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid beta (Abeta) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J. Biol. Chem. 2012, 287, 8154–8162. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef]
- Yun, S.; Yun, S.; Guy, H.R. Analysis of the stabilities of hexameric amyloid-beta(1–42) models using discrete molecular dynamics simulations. J. Mol. Graph. Model. 2011, 29, 657–662. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef]
- Parsons, C.G.; Ruitenberg, M.; Freitag, C.E.; Sroka-Saidi, K.; Russ, H.; Rammes, G. MRZ-99030—A novel modulator of Abeta aggregation: I—Mechanism of action (MoA) underlying the potential neuroprotective treatment of Alzheimer’s disease, glaucoma and age-related macular degeneration (AMD). Neuropharmacology 2015, 92, 158–169. [Google Scholar] [CrossRef]
- Rammes, G.; Parsons, C.G. The Abeta aggregation modulator MRZ-99030 prevents and even reverses synaptotoxic effects of Abeta(1–42) on LTP even following serial dilution to a 500:1 stoichiometric excess of Abeta(1–42), suggesting a beneficial prion-like seeding mechanism. Neuropharmacology 2020, 179, 108267. [Google Scholar] [CrossRef]
- Russ, H.; Danysz, W.; Parsons, C.G. Interval Therapy for the Treatment of Loss of Eyesight in Humans with Glaucoma and Other Degenerative Eye Diseases. Patent WO2013189606A3, 27 December 2013. [Google Scholar]
- Russ, H.; Mazzanti, M.; Parsons, C.; Riemann, K.; Gebauer, A.; Rammes, G. The Small Molecule GAL-201 Efficiently Detoxifies Soluble Amyloid beta Oligomers: New Approach towards Oral Disease-Modifying Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 5794. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-beta and alpha-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Younkin, S.G. The role of A beta 42 in Alzheimer’s disease. J. Physiol. Paris 1998, 92, 289–292. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Mahil, D.S.; Patel, B.P.; Austen, B.M. Oligomerization and toxicity of beta-amyloid-42 implicated in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2000, 273, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Violani, E.; Salis, S.; Venezia, V.; Dolcini, V.; Damonte, G.; Benatti, U.; D’Arrigo, C.; Patrone, E.; Carlo, P.; et al. Pyroglutamate-modified amyloid beta-peptides--AbetaN3(pE)--strongly affect cultured neuron and astrocyte survival. J. Neurochem. 2002, 82, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; Konig, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef]
- Risher, W.C.; Ustunkaya, T.; Singh Alvarado, J.; Eroglu, C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE 2014, 9, e107591. [Google Scholar] [CrossRef]
- Kater, S.B.; Rehder, V. The sensory-motor role of growth cone filopodia. Curr. Opin. Neurobiol. 1995, 5, 68–74. [Google Scholar] [CrossRef]
- Ziv, N.E.; Smith, S.J. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 1996, 17, 91–102. [Google Scholar] [CrossRef]
- Nimchinsky, E.A.; Sabatini, B.L.; Svoboda, K. Structure and function of dendritic spines. Annu. Rev. Physiol. 2002, 64, 313–353. [Google Scholar] [CrossRef]
- Bourne, J.; Harris, K.M. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 2007, 17, 381–386. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Houston, M.E., Jr.; Campbell, A.P.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hodges, R.S. Lactam bridge stabilization of alpha-helices: The role of hydrophobicity in controlling dimeric versus monomeric alpha-helices. Biochemistry 1996, 35, 10041–10050. [Google Scholar] [CrossRef]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Overman, M.J.; Cotman, C.W. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J. Biol. Chem. 1995, 270, 23895–23898. [Google Scholar] [CrossRef] [PubMed]
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Abeta42 Anti-Aggregation Mechanism of Action of Tramiprosate in Alzheimer’s Disease: Integrating Molecular Analytical Methods, Pharmacokinetic and Clinical Data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y. Molecular mechanism of hippocampal long-term potentiation—Towards multiscale understanding of learning and memory. Neurosci. Res. 2022, 175, 3–15. [Google Scholar] [CrossRef]
- Yuste, R.; Bonhoeffer, T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu. Rev. Neurosci. 2001, 24, 1071–1089. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Ellis-Davies, G.C.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef]
- Han, X.; Liu, Y.J.; Liu, B.W.; Ma, Z.L.; Xia, T.J.; Gu, X.P. TREM2 and CD163 Ameliorate Microglia-Mediated Inflammatory Environment in the Aging Brain. J. Mol. Neurosci. 2022, 72, 1075–1084. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernandez, L.; Garcia-Dominguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Okuzono, Y.; Sakuma, H.; Miyakawa, S.; Ifuku, M.; Lee, J.; Das, D.; Banerjee, A.; Zhao, Y.; Yamamoto, K.; Ando, T.; et al. Reduced TREM2 activation in microglia of patients with Alzheimer’s disease. FEBS Open Bio 2021, 11, 3063–3080. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Diaz-Lucena, D.; Ainciburu, M.; Torrejon-Escribano, B.; Carmona, M.; Llorens, F.; Ferrer, I. Glutamate Transporter GLT1 Expression in Alzheimer Disease and Dementia With Lewy Bodies. Front. Aging Neurosci. 2018, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Vigo-Pelfrey, C.; Teplow, D.B.; Miller, C.; Schenk, D.; Johnston, J.; Winblad, B.; Venizelos, N.; Lannfelt, L.; Selkoe, D.J. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl. Acad. Sci. USA 1994, 91, 11993–11997. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.B.; Frerking, M.; Zhou, Q. Spine expansion and stabilization associated with long-term potentiation. J. Neurosci. 2008, 28, 5740–5751. [Google Scholar] [CrossRef]
- Lang, C.; Barco, A.; Zablow, L.; Kandel, E.R.; Siegelbaum, S.A.; Zakharenko, S.S. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 16665–16670. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Galatro, T.F.; Vainchtein, I.D.; Brouwer, N.; Boddeke, E.; Eggen, B.J.L. Isolation of Microglia and Immune Infiltrates from Mouse and Primate Central Nervous System. Methods Mol. Biol. 2017, 1559, 333–342. [Google Scholar] [CrossRef]
- Clark, I.C.; Wheeler, M.A.; Lee, H.G.; Li, Z.; Sanmarco, L.M.; Thaploo, S.; Polonio, C.M.; Shin, S.W.; Scalisi, G.; Henry, A.R.; et al. Identification of astrocyte regulators by nucleic acid cytometry. Nature 2023, 614, 326–333. [Google Scholar] [CrossRef] [PubMed]
- de Haas, A.H.; Boddeke, H.W.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Gorina, Y.V.; Salmina, A.B.; Erofeev, A.I.; Gerasimov, E.I.; Bolshakova, A.V.; Balaban, P.M.; Bezprozvanny, I.B.; Vlasova, O.L. Astrocyte Activation Markers. Biochemistry 2022, 87, 851–870. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Gomez-Isla, T.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol. 2013, 182, 2332–2344. [Google Scholar] [CrossRef]
- Kleinknecht, K.R.; Bedenk, B.T.; Kaltwasser, S.F.; Grunecker, B.; Yen, Y.C.; Czisch, M.; Wotjak, C.T. Hippocampus-dependent place learning enables spatial flexibility in C57BL6/N mice. Front. Behav. Neurosci. 2012, 6, 87. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Neumuller, T.; Klug, C.; Fuchs, S.; Schlegel, M.; Ballmann, M.; Tartler, K.J.; Pianos, A.; Garcia, M.S.; Liere, P.; et al. Chronic administration of XBD173 ameliorates cognitive deficits and neuropathology via 18 kDa translocator protein (TSPO) in a mouse model of Alzheimer’s disease. Transl. Psychiatry 2023, 13, 332. [Google Scholar] [CrossRef]
- Rothman, K.J. No adjustments are needed for multiple comparisons. Epidemiology 1990, 1, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Hentschke, H.; Stuttgen, M.C. Computation of measures of effect size for neuroscience data sets. Eur. J. Neurosci. 2011, 34, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | SD5 (20 min) | SD5 (1 h) | SD5 (1 d) | SD5 (1 w) | |
|---|---|---|---|---|---|
| Aβ1–42 | p < 0.0001 with AUC = 0 [0; 0] | - | AUC = 0.13 [0; 0.33] | AUC = 0.16 [0; 0.42] | AUC = 0.09 [0; 0.28] |
| Aβ1–40 | p < 0.001 with AUC = 0 [0; 0] | p < 0.05 with AUC = 0.06 [0; 0.2] | AUC = 0.08 [0; 0.28] | AUC = 0.1719 [0; 0.44] | p < 0.01 with AUC = 0.02 [0; 0.09] |
| AβpE3 | p < 0.0001 with AUC = 0 [0; 0] | - | p < 0.05 with AUC = 0.08 [0; 0.25] | AUC = 0.13 [0; 0.38] | AUC = 0.23 [0; 0.53] |
| 3NTyr(10)-Aβ | p < 0.01 with AUC = 0 [0; 0] | p < 0.05 with AUC = 0.08 [0; 0.25] | AUC = 0.17 [0; 0.43] | p < 0.05 with AUC = 0.13 [0; 0.34] | p < 0.05 with AUC = 0.06 [0; 0.23] |
| Control Naive | Control Naive LTP | SD5 (20 min) | SD5 (1 h) | SD5 (1 d) | SD5 (1 w) | |
|---|---|---|---|---|---|---|
| Aβ1–42 | AUC = 0.11 [0; 0.31] | p < 0.05 with AUC = 0.10 [0; 0.33] | p < 0.01 with AUC = 0.05 [0; 0.19] | AUC = 0.22 [0.03; 0.48] | AUC = 0.31 [0.08; 0.59] | AUC = 0.27 [0.04; 0.57] |
| Aβ1–40 | p < 0.05 with AUC = 0.05 [0; 0.19] | AUC = 0.08 [0; 0.31] | AUC = 0.17 [0; 0.44] | AUC = 0.16 [0; 0.41] | AUC = 0.35 [0.08; 0.69] | p < 0.01 with AUC = 0.02 [0; 0.09] |
| AβpE3 | p < 0.05 with AUC = 0.06 [0; 0.23] | AUC = 0.06 [0; 0.25] | AUC = 0.11 [0; 0.33] | AUC = 0.13 [0; 0.36] | p < 0.05 with AUC = 0.05 [0; 0.19] | p < 0.001 with AUC = 0.03 [0; 0.14] |
| 3NTyr(10)-Aβ | AUC = 0.17 [0; 0.42] | p < 0.05 with AUC = 0.17 [0; 0.46] | AUC = 0.20 [0; 0.47] | AUC = 0.22 [0.03; 0.47] | AUC = 0.17 [0; 0.41] | AUC = 0.48 [0.19; 0.78] |
| Control Naive | Control Naive LTP | SD5 (20 min) | SD5 (1 h) | SD5 (1 d) | SD5 (1 w) | |
|---|---|---|---|---|---|---|
| Filopodia | AUC = 0.77 [0.56; 0.94] | AUC = 0.81 [0.63; 0.94] | AUC = 0.71 [0.47; 0.94] | AUC = 0.82 [0.56; 1] | AUC = 0.68 [0.41; 0.91] | AUC = 0.5 [0.2; 0.8] |
| Long-Thin | AUC = 0.9 [0.7; 1] | AUC = 0.84 [0.58; 1] | AUC = 0.45 [0.17; 0.77] | AUC = 0.82 [0.56; 1] | AUC = 0.53 [0.23; 0.83] | AUC = 0.57 [0.25; 0.88] |
| Thin | AUC = 0.56 [0.25; 0.84] | AUC = 0.4 [0.13; 0.73] | AUC = 0.19 [0.02; 0.44] | p < 0.05 with AUC = 0.14 [0; 0.36] | AUC = 0.2 [0.02; 0.47] | AUC = 0.32 [0.07; 0.63] |
| Stubby | p < 0.05 with AUC = 0.08 [0; 0.23] | AUC = 0.04 [0; 0.17] | AUC = 0.41 [0.13; 0.7] | AUC = 0.27 [0.05; 0.55] | AUC = 0.61 [0.3; 0.89] | AUC = 0.39 [0.07; 0.73] |
| Mushroom | AUC = 0.05 [0; 0.17] | AUC = 0.54 [0.21; 0.88] | AUC = 0.41 [0.13; 0.7] | AUC = 0.84 [0.61; 1] | AUC = 0.84 [0.59; 1] | AUC = 0.59 [0.27; 0.89] |
| Control | SD5 (20 min) | SD5 (1 h) | SD5 (1 d) | SD5 (1 w) | |
|---|---|---|---|---|---|
| Relative count microglia | AUC = 0.5 [0.2; 0.81] | p < 0.05 with AUC = 0.09 [0; 0.28] | p < 0.01 with AUC = 0.03 [0; 0.14] | p < 0.001 with AUC = 0 [0; 0] | AUC = 0.01 [0; 0.05] |
| CD163 | p < 0.05 with AUC = 0 [0; 0] | p < 0.05 with AUC = 0 [0; 0] | p < 0.01 with AUC = 0 [0; 0] | p < 0.0001 with AUC = 0 [0; 0] | p < 0.001 with AUC = 0 [0; 0] |
| TREM-2 | p < 0.05 with AUC = 0.06 [0; 0.22] | AUC = 0.22 [0.03; 0.48] | AUC = 0.11 [0; 0.31] | AUC = 0.16 [0; 0.39] | p < 0.05 with AUC = 0.05 [0; 0.19] |
| IL-1β | AUC = 0.86 [0.63; 1] | p < 0.0001 with AUC 0.98 [0.91; 1] | p < 0.001 with AUC = 0.97 [0.86; 1] | p < 0.05 with AUC = 0.94 [0.77; 1] | p < 0.05 with AUC = 0.95 [0.81; 1] |
| Relative count reactive astrocytes | p < 0.05 with AUC = 0.86 [0.64; 1] | p < 0.01 with AUC = 1 [1; 1] | p < 0.001 with AUC = 1 [1; 1] | AUC = 1 [1; 1] | p < 0.05 with AUC = 1 [1; 1] |
| ALDH1L1 | p < 0.05 with AUC = 1 [1; 1] | p < 0.001 with AUC = 1 [1; 1] | AUC = 0.98 [0.93; 1] | p < 0.05 with AUC = 0.89 [0.67; 1] | p < 0.01 with AUC = 1 [1; 1] |
| CD45 | Brilliant Violet 510™ anti-mouse CD45, BioLegend (San Diego, CA, USA), CAT: 103137 |
| CD11b | Brilliant Violet 785™ anti-mouse/human CD11b, BioLegend (San Diego, CA, USA), CAT: 101243 |
| F4/80 | PerCP/Cyanine5.5 anti-mouse F4/80, BioLegend (San Diego, CA, USA), CAT: 123127 |
| TMEM119 | Anti-Mo, TMEM119 PE_Cyanine7, Invitrogen (Waltham, MA, USA), CAT: 25-6119-80 |
| P2RY12 | APC anti-P2RY12, BioLegend (San Diego, CA, USA), CAT: 848005 |
| CD163 | Anti-Mo CD163 SuperBright™ 702, Invitrogen (Waltham, MA, USA), CAT: 67-1631-82 |
| TREM-2 | TREM-2 Monoclonal Antibody FITC, Invitrogen (Waltham, MA, USA), CAT: MA5-28223 |
| IL-1β | IL-1Beta Rabbit anti-Mouse Polyclonal (aa118-269) (APC, Cy7) Antibody, LS Bio (Lynnwood, WA, USA), CAT: LS-C730762-100 |
| GFAP | Mouse anti-GFAP (SFM248) [DyLight 594], Novus Biologicals (Centennial, CO, USA), CAT: NBP2-34401DL594 |
| ALDH1L1 | ALDH1L1 Antibody (2E7) [DyLight 680], Novus Biologicals (Centennial, CO, USA), CAT: NBP2-50033FR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riemann, K.; von Ahsen, J.; Böhm, T.; Schlegel, M.; Kreuzer, M.; Fenzl, T.; Russ, H.; Parsons, C.G.; Rammes, G. GAL-201 as a Promising Amyloid-β-Targeting Small-Molecule Approach for Alzheimer’s Disease Treatment: Consistent Effects on Synaptic Plasticity, Behavior and Neuroinflammation. Int. J. Mol. Sci. 2025, 26, 4167. https://doi.org/10.3390/ijms26094167
Riemann K, von Ahsen J, Böhm T, Schlegel M, Kreuzer M, Fenzl T, Russ H, Parsons CG, Rammes G. GAL-201 as a Promising Amyloid-β-Targeting Small-Molecule Approach for Alzheimer’s Disease Treatment: Consistent Effects on Synaptic Plasticity, Behavior and Neuroinflammation. International Journal of Molecular Sciences. 2025; 26(9):4167. https://doi.org/10.3390/ijms26094167
Chicago/Turabian StyleRiemann, Katrin, Jeldrik von Ahsen, Tamara Böhm, Martin Schlegel, Matthias Kreuzer, Thomas Fenzl, Hermann Russ, Christopher G. Parsons, and Gerhard Rammes. 2025. "GAL-201 as a Promising Amyloid-β-Targeting Small-Molecule Approach for Alzheimer’s Disease Treatment: Consistent Effects on Synaptic Plasticity, Behavior and Neuroinflammation" International Journal of Molecular Sciences 26, no. 9: 4167. https://doi.org/10.3390/ijms26094167
APA StyleRiemann, K., von Ahsen, J., Böhm, T., Schlegel, M., Kreuzer, M., Fenzl, T., Russ, H., Parsons, C. G., & Rammes, G. (2025). GAL-201 as a Promising Amyloid-β-Targeting Small-Molecule Approach for Alzheimer’s Disease Treatment: Consistent Effects on Synaptic Plasticity, Behavior and Neuroinflammation. International Journal of Molecular Sciences, 26(9), 4167. https://doi.org/10.3390/ijms26094167