
The Role of RAS in CNS Tumors: A Key Player or an Overlooked Oncogene?

 , , and
, , and
Abstract
1. Introduction
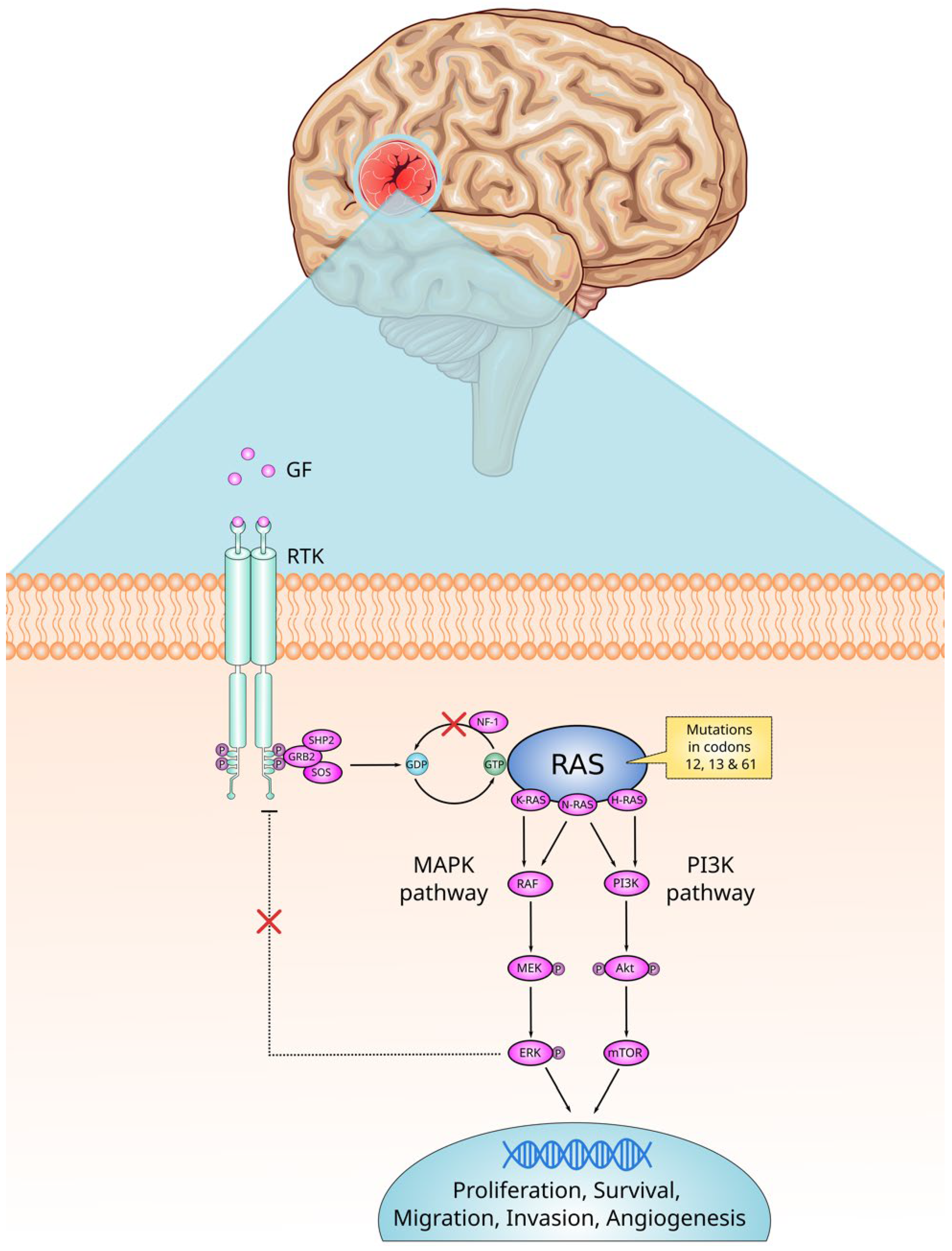
2. RAS Signaling
3. RAS and the CNS
4. Indirect Activation of RAS in CNS Tumors
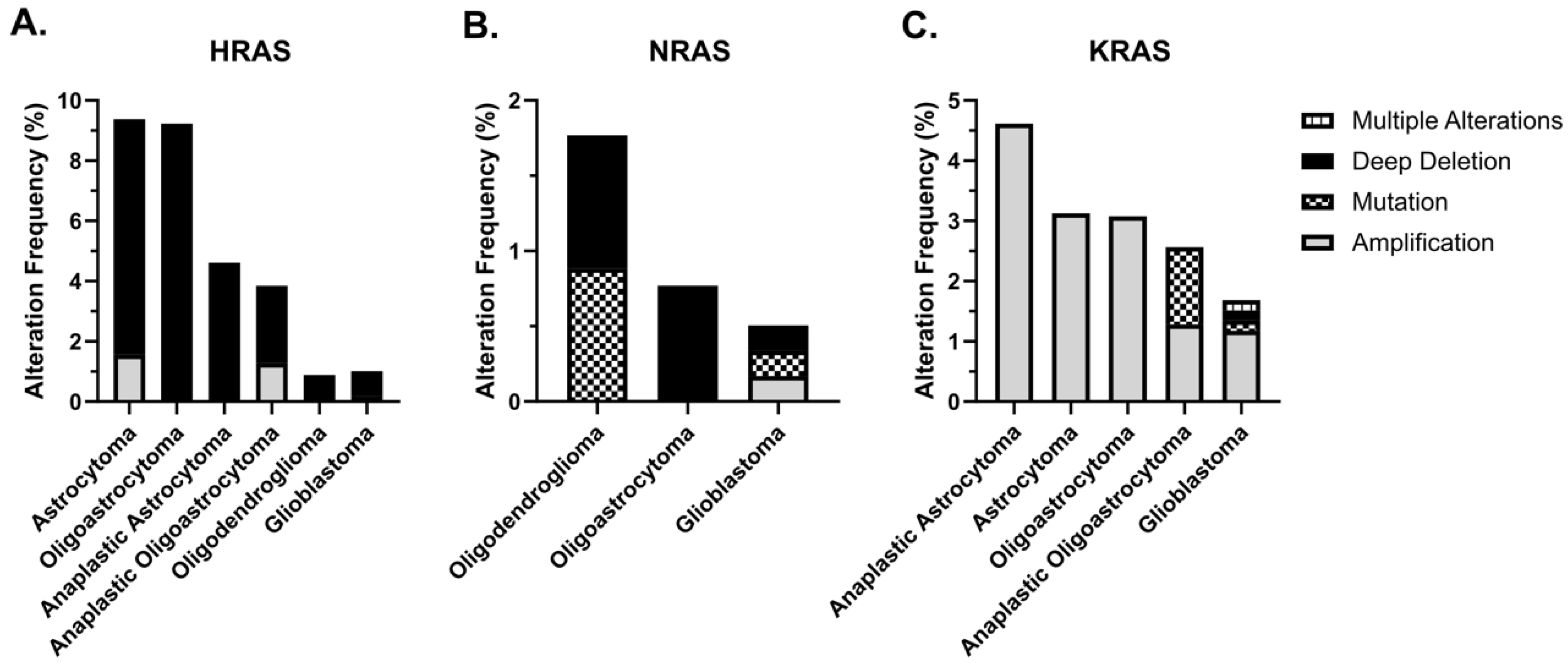
5. KRAS and Glioblastoma
6. Strategies to Inhibit Mutated RAS
6.1. Disrupting Post-Translational Lipid Modifications at the RAS C-Terminal
6.2. Inhibitors of the RAS–Effector Interaction
6.3. Covalent Inhibition of KRASG12C
6.4. Current Clinical Trials Involving CNS Tumors and RAS
7. Materials and Methods
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Yuan, L.; Lin, A.; Lin, H.; Huang, X.; Ruan, J.; Zhuo, Z. KRAS gene polymorphisms are associated with the risk of glioma: A two-center case-control study. Transl. Pediatr. 2021, 10, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Parada, L.F.; Tabin, C.J.; Shih, C.; Weinberg, R.A. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 1982, 297, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of Ras mutations in cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Denayer, E.; de Ravel, T.; Legius, E. Clinical and molecular aspects of RAS related disorders. J. Med. Genet. 2008, 45, 695–703. [Google Scholar] [CrossRef]
- Aran, V. K-RAS4A: Lead or Supporting Role in Cancer Biology? Front. Mol. Biosci. 2021, 8, 729830. [Google Scholar] [CrossRef]
- Aran, V.; Zalis, M.; Montella, T.; de Sousa, C.A.M.; Ferrari, B.L.; Gil Ferreira, C. Evaluation of KRAS Concomitant Mutations in Advanced Lung Adenocarcinoma Patients. Med. Kaunas Lith. 2021, 57, 1039. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Makino, Y.; Arakawa, Y.; Yoshioka, E.; Shofuda, T.; Minamiguchi, S.; Kawauchi, T.; Tanji, M.; Kanematsu, D.; Nonaka, M.; Okita, Y.; et al. Infrequent RAS mutation is not associated with specific histological phenotype in gliomas. BMC Cancer 2021, 21, 1025. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRASG12C inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Laude, A.J.; Prior, I.A. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J. Cell Sci. 2008, 121, 421–427. [Google Scholar] [CrossRef]
- Plowman, S.J.; Williamson, D.J.; O’Sullivan, M.J.; Doig, J.; Ritchie, A.-M.; Harrison, D.J.; Melton, D.W.; Arends, M.J.; Hooper, M.L.; Patek, C.E. While K-ras Is Essential for Mouse Development, Expression of the K-ras 4A Splice Variant Is Dispensable. Mol. Cell. Biol. 2003, 23, 9245–9250. [Google Scholar] [CrossRef]
- Chen, W.-C.; To, M.D.; Westcott, P.M.K.; Delrosario, R.; Kim, I.-J.; Philips, M.; Tran, Q.; Bayani, N.; Balmain, A. Regulation of KRAS4A/B splicing in cancer stem cells by the RBM39 splicing complex. bioRxiv 2019. [Google Scholar] [CrossRef]
- Wellbrock, C.; Arozarena, I. The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Front. Cell Dev. Biol. 2016, 4, 33. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- AKT/PKB Signaling: Navigating the Network: Cell. Available online: https://www.cell.com/cell/fulltext/S0092-8674(17)30413-0 (accessed on 16 April 2025).
- Durand-Onaylı, V.; Haslauer, T.; Härzschel, A.; Hartmann, T.N. Rac GTPases in Hematological Malignancies. Int. J. Mol. Sci. 2018, 19, 4041. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Sullivan, A.J.; Medina, R.; Ito, Y.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004, 23, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Funa, K.; Sasahara, M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2014, 9, 168–181. [Google Scholar] [CrossRef]
- Di Liberto, V.; Mudò, G.; Belluardo, N. Crosstalk between receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCR) in the brain: Focus on heteroreceptor complexes and related functional neurotrophic effects. Neuropharmacology 2019, 152, 67–77. [Google Scholar] [CrossRef]
- Sheffels, E.; Kortum, R.L. The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes 2021, 12, 662. [Google Scholar] [CrossRef]
- To, M.D.; Rosario, R.D.; Westcott, P.M.K.; Banta, K.L.; Balmain, A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene 2013, 32, 4028–4033. [Google Scholar] [CrossRef]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Mokhtari, K.; Duyckaerts, C. The 2007 WHO classification of tumors of the central nervous system—What has changed? Curr. Opin. Neurol. 2008, 21, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Kothari, S.; Rahman, R.; Lee, E.Q.; Dunn, G.P.; Galanis, E.; Chang, S.M.; Nabors, L.B.; Ahluwalia, M.S.; Stupp, R.; et al. Glioblastoma Clinical Trials: Current Landscape and Opportunities for Improvement. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Messina, S. The RAS oncogene in brain tumors and the involvement of let-7 microRNA. Mol. Biol. Rep. 2024, 51, 531. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics *. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Guha, A.; Feldkamp, M.M.; Lau, N.; Boss, G.; Pawson, A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene 1997, 15, 2755–2765. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Q.; Kutlu, B.; Difilippantonio, S.; Bash, R.; Gilbert, D.; Yin, C.; O’Sullivan, T.N.; Yang, C.; Kozlov, S.; et al. Evolutionary etiology of high-grade astrocytomas. Proc. Natl. Acad. Sci. USA 2013, 110, 17933–17938. [Google Scholar] [CrossRef]
- Wiestler, O.D.; Brüstle, O.; Eibl, R.H.; Radner, H.; Von Deimling, A.; Plate, K.; Aguzzi, A.; Kleihues, P. A new approach to the molecular basis of neoplastic transformation in the brain. Neuropathol. Appl. Neurobiol. 1992, 18, 443–453. [Google Scholar] [CrossRef]
- Schittenhelm, J.; Krischker, N.; Gepfner-Tuma, I.; Behling, F.; Noell, S.; Eckert, F.; Biskup, S.; Tabatabai, G. Oncogenic KRAS hotspot mutations are rare in IDH-mutant gliomas. Brain Pathol. 2019, 29, 321–324. [Google Scholar] [CrossRef]
- Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Jahromi, B.R.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.-R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 mutations at residue p.R132 (IDH1R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Chi, A.S.; Batchelor, T.T.; Dias-Santagata, D.; Borger, D.; Stiles, C.D.; Wang, D.L.; Curry, W.T.; Wen, P.Y.; Ligon, K.L.; Ellisen, L.; et al. Prospective, high-throughput molecular profiling of human gliomas. J. Neurooncol. 2012, 110, 89–98. [Google Scholar] [CrossRef]
- Pekmezci, M.; Villanueva-Meyer, J.E.; Goode, B.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Bastian, B.C.; Chamyan, G.; Maher, O.M.; Khatib, Z.; et al. The genetic landscape of ganglioglioma. Acta Neuropathol. Commun. 2018, 6, 47. [Google Scholar] [CrossRef]
- Brüstle, O.; Ohgaki, H.; Schmitt, H.P.; Walter, G.F.; Ostertag, H.; Kleihues, P. Primitive neuroectodermal tumors after prophylactic central nervous system irradiation in children. Association with an activated K-ras gene. Cancer 1992, 69, 2385–2392. [Google Scholar] [CrossRef]
- Aran, V.; Heringer, M.; da Mata, P.J.; Kasuki, L.; Miranda, R.L.; Andreiuolo, F.; Chimelli, L.; Filho, P.N.; Gadelha, M.R.; Neto, V.M. Identification of mutant K-RAS in pituitary macroadenoma. Pituitary 2021, 24, 746–753. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Langdon, J.A.; Hollander, A.; Hernan, R.; Hogg, T.L.; Gajjar, A.; Fuller, C.; Clifford, S.C. Mutational analysis of PDGFR-RAS/MAPK pathway activation in childhood medulloblastoma. Eur. J. Cancer Oxf. Engl. 1990 2006, 42, 646–649. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain Tumor Mutations Detected in Cerebral Spinal Fluid. Clin. Chem. 2015, 61, 514. [Google Scholar] [CrossRef]
- Rabbie, R.; Ferguson, P.; Wong, K.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; Lakhani, S.R.; et al. Hotspot KRAS mutations in brain metastases at the first metastatic recurrence of cutaneous melanoma. Br. J. Cancer 2021, 124, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Swinkels, D.W.; de Kok, J.B.; Hanselaar, A.; Lamers, K.; Boerman, R.H. Early Detection of Leptomeningeal Metastasis by PCR Examination of Tumor-derived K-ras DNA in Cerebrospinal Fluid. Clin. Chem. 2000, 46, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Wu, Y.; Ma, X.; Yan, Y.; Shao, S.; Liu, J.; Ma, H.; Liu, R.; Chai, L.; Ren, J. A Comprehensive Meta-Analysis of Association between EGFR Mutation Status and Brain Metastases in NSCLC. Pathol. Oncol. Res. POR 2019, 25, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.R.; Likhacheva, A.; Pinnix, C.; Allen, P.K.; Prabhu, S.S.; Guha-Thakurta, N.; Welsh, J.W.; Brown, P.D.; Chang, E.L. Prognostic significance of EGFR and KRAS mutations in NSCLC patients with brain metastases treated with radiosurgery. J. Radiosurgery SBRT 2015, 3, 171–178. [Google Scholar]
- Lauko, A.; Kotecha, R.; Barnett, A.; Li, H.; Tatineni, V.; Ali, A.; Patil, P.; Mohammadi, A.M.; Chao, S.T.; Murphy, E.S.; et al. Impact of KRAS mutation status on the efficacy of immunotherapy in lung cancer brain metastases. Sci. Rep. 2021, 11, 18174. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, S.; Zhang, Q.; Li, S.; Ren, D.; Ren, F.; Zu, L.; Wang, Y.; Lei, X.; Zhou, N.; et al. Complete disease remission in a TP53 and KRAS co-mutated brain oligometastatic lung cancer patient after immuno-chemotherapy and surgical resection: A case report. Transl. Lung Cancer Res. 2021, 10, 2298–2305. [Google Scholar] [CrossRef]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzàlez-Juncà, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef]
- Wang, X.-F.; Shi, Z.-M.; Wang, X.-R.; Cao, L.; Wang, Y.-Y.; Zhang, J.-X.; Yin, Y.; Luo, H.; Kang, C.-S.; Liu, N.; et al. MiR-181d acts as a tumor suppressor in glioma by targeting K-ras and Bcl-2. J. Cancer Res. Clin. Oncol. 2012, 138, 573–584. [Google Scholar] [CrossRef]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Zhao, Y.; Kang, J.-H.; Yoo, K.-C.; Kang, S.-G.; Lee, H.-J.; Lee, S.-J. K-RAS Acts as a Critical Regulator of CD44 to Promote the Invasiveness and Stemness of GBM in Response to Ionizing Radiation. Int. J. Mol. Sci. 2021, 22, 10923. [Google Scholar] [CrossRef] [PubMed]
- Brock, E.J.; Ji, K.; Reiners, J.J.; Mattingly, R.R. How to Target Activated Ras Proteins: Direct Inhibition vs. Induced Mislocalization. Mini Rev. Med. Chem. 2016, 16, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Spyris, C.D.; Castellino, R.C.; Schniederjan, M.J.; Kadom, N. High-Grade Gliomas in Children with Neurofibromatosis Type 1: Literature Review and Illustrative Cases. AJNR Am. J. Neuroradiol. 2019, 40, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Park, G.-H.; Lee, S.-J.; Lee, C.-G.; Kim, J.; Park, E.; Jeong, S.-Y. Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor. Int. J. Mol. Sci. 2021, 22, 13308. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef]
- Magalhaes, Y.T.; Boell, V.K.; Cardella, G.D.; Forti, F.L. Downregulation of the Rho GTPase pathway abrogates resistance to ionizing radiation in wild-type p53 glioblastoma by suppressing DNA repair mechanisms. Cell Death Dis. 2023, 14, 283. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Chakrabarti, I.; Cockburn, M.; Cozen, W.; Wang, Y.-P.; Preston-Martin, S. A population-based description of glioblastoma multiforme in Los Angeles County, 1974–1999. Cancer 2005, 104, 2798–2806. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Blomquist, M.R.; Ensign, S.F.; D’Angelo, F.; Phillips, J.J.; Ceccarelli, M.; Peng, S.; Halperin, R.F.; Caruso, F.P.; Garofano, L.; Byron, S.A.; et al. Temporospatial genomic profiling in glioblastoma identifies commonly altered core pathways underlying tumor progression. Neuro-Oncology Adv. 2020, 2, vdaa078. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Zhao, J.; Jung, S.W.; Ladewig, E.; Kong, D.-S.; Suh, Y.-L.; Lee, Y.; Kim, D.; Ahn, S.H.; Bordyuh, M.; et al. Distinct genomic profile and specific targeted drug responses in adult cerebellar glioblastoma. Neuro-Oncology 2019, 21, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Reifenberger, J.; Reifenberger, G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004, 108, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Zuchegna, C.; Leone, S.; Romano, A.; Porcellini, A.; Messina, S. KRAS is a molecular determinant of platinum responsiveness in glioblastoma. BMC Cancer 2024, 24, 77. [Google Scholar] [CrossRef]
- Steele, V.E.; Lubet, R.A. The Use of Animal Models for Cancer Chemoprevention Drug Development. Semin. Oncol. 2010, 37, 327–338. [Google Scholar] [CrossRef]
- Holmen, S.L.; Williams, B.O. Essential Role for Ras Signaling in Glioblastoma Maintenance. Cancer Res. 2005, 65, 8250–8255. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the Regulator: Post-Translational Modification of Ras. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Takashima, A.; Faller, D.V. Targeting the RAS oncogene. Expert Opin. Ther. Targets 2013, 17, 507–531. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D.; Tan, B.R.; Khan, G.N.; Zalupski, M.M.; et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer 2017, 116, 575. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a potent and selective SOS1::KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Gambella, A.; Senetta, R.; Collemi, G.; Vallero, S.G.; Monticelli, M.; Cofano, F.; Zeppa, P.; Garbossa, D.; Pellerino, A.; Rudà, R.; et al. NTRK Fusions in Central Nervous System Tumors: A Rare, but Worthy Target. Int. J. Mol. Sci. 2020, 21, 753. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Cocco, E.; Schram, A.M.; Kulick, A.; Misale, S.; Won, H.H.; Yaeger, R.; Razavi, P.; Ptashkin, R.; Hechtman, J.F.; Toska, E.; et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med. 2019, 25, 1422–1427. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Zahra, K.; Shabbir, M.; Badshah, Y.; Trembley, J.H.; Badar, Z.; Khan, K.; Afsar, T.; Almajwal, A.; Alruwaili, N.W.; Razak, S. Determining KLF14 tertiary structure and diagnostic significance in brain cancer progression. Sci. Rep. 2022, 12, 8039. [Google Scholar] [CrossRef] [PubMed]
- Merulla, A.E.; Stella, M.; Barbagallo, C.; Battaglia, R.; Caponnetto, A.; Broggi, G.; Altieri, R.; Certo, F.; Caltabiano, R.; Ragusa, M.; et al. circSMARCA5 Is an Upstream Regulator of the Expression of miR-126-3p, miR-515-5p, and Their mRNA Targets, Insulin-like Growth Factor Binding Protein 2 (IGFBP2) and NRAS Proto-Oncogene, GTPase (NRAS) in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 13676. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent small molecule pan-RAS inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Xie, R.; Huang, H.; Chen, T.; Huang, X.; Chen, C. Effectiveness and safety of pelareorep plus chemotherapy versus chemotherapy alone for advanced solid tumors: A meta-analysis. Front. Pharmacol. 2023, 14, 1228225. [Google Scholar] [CrossRef]
- Neonc Technologies, Inc. An Open-Label Phase 1/2 Dose Finding, Safety and Efficacy Study of Oral Neo212 in Patients with Astrocytoma Idh-Mutant, Glioblastoma Idh-Wildtype or Uncontrolled Brain Metastasis in Patients with Select Solid Tumors. Clinicaltrials.gov. Report No.: NCT06047379. 2024. Available online: https://clinicaltrials.gov/study/NCT06047379 (accessed on 31 December 2023).
- Maastricht University Medical Center. A Phase II Study Evaluating Intracranial Efficacy of JDQ443 in Patients with KRAS G12C+ NSCLC and Brain Metastases [Internet]. Clinicaltrials.gov. Report No.: NCT05999357. March 2024. Available online: https://clinicaltrials.gov/study/NCT05999357 (accessed on 31 December 2023).
- Criterium, Inc. A Phase I/II Study of AMG 510 in Combination with MVASI in Patients with Advanced, Unresectable or Meta-static KRAS G12C Mutant NSCLC with Asymptomatic Brain Metastasis [Internet]. Clinicaltrials.gov. Report No.: NCT05180422. October 2023. Available online: https://clinicaltrials.gov/study/NCT05180422 (accessed on 31 December 2023).
- Gentzler, R. Phase II Study of Adagrasib + Stereotactic Radiosurgery (SRS) for Patients with Metastatic KRAS G12C-Mutated NSCLC with Untreated Brain Metastases [Internet]. Clinicaltrials.gov. Report No.: NCT06248606. January 2024. Available online: https://clinicaltrials.gov/study/NCT06248606 (accessed on 31 December 2023).
- Black Diamond Therapeutics, Inc. A Phase 1, Open-Label Study of Oral BDTX-4933 in Patients with KRAS, BRAF and Other Select RAS/MAPK Mutation Positive Neoplasms [Internet]. Clinicaltrials.gov. Report No.: NCT05786924. February 2024. Available online: https://clinicaltrials.gov/study/NCT05786924 (accessed on 31 December 2023).
- Wakelee, H. Study of Regorafenib in Combination with Oral Methotrexate for KRAS Mutated Non-Small Cell Lung Cancer (NSCLC) [Internet]. Clinicaltrials.gov. Report No.: NCT03520842. July 2023. Available online: https://clinicaltrials.gov/study/NCT03520842 (accessed on 31 December 2023).
- Alliance for Clinical Trials in Oncology. Genomically-Guided Treatment Trial in Brain Metastases [Internet]. Clinicaltrials.gov. Report No.: NCT03994796. February 2024. Available online: https://clinicaltrials.gov/study/NCT03994796 (accessed on 31 December 2023).
- xCures. Expanded Access to Ulixertinib (BVD-523) in Patients with Advanced MAPK Pathway-Altered Malignancies [Internet]. Clinicaltrials.gov. Report No.: NCT04566393. August 2023. Available online: https://clinicaltrials.gov/study/NCT04566393 (accessed on 31 December 2023).
- Safety and Efficacy of NEO212 in Patients with Astrocytoma IDH-mutant, Glioblastoma IDH-wildtype or Brain Metastasis—NCI. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2024-08760 (accessed on 6 February 2025).
- Study of Efficacy and Safety of Dabrafenib in Combination with Trametinib in Pediatric Patients with BRAF V600 Mutation Positive LGG or Relapsed or Refractory HGG Tumors—NCI. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCT02684058 (accessed on 6 February 2025).
- A Study to Determine Safety, Tolerability and Pharmacokinetics of Oral Dabrafenib In Children and Adolescent Subjects—NCI. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2013-00631 (accessed on 6 February 2025).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Trial Name | Therapy Used | ID | Reference |
|---|---|---|---|
| A Phase II Study evaluating intracranial efficacy of JDQ443 in Patients With KRAS G12C+ NSCLC and Brain Metastases |
| NCT05999357 | [101] |
| A Phase I/II Study of AMG 510 in Combination with MVASI in Patients with Advanced, Unresectable or Metastatic KRAS G12C Mutant NSCLC With Asymptomatic Brain Metastasis. |
| NCT05180422 | [102] |
| Phase II Study of Adagrasib + Stereotactic Radiosurgery (SRS) for Patients with Metastatic KRAS G12C-mutated NSCLC With Untreated Brain Metastases |
| NCT06248606 | [103] |
| A Phase 1, Open-label Study of Oral BDTX-4933 in Patients With KRAS, BRAF and Other Select RAS/MAPK Mutation Positive Neoplasms |
| NCT05786924 | [104] |
| Study of Regorafenib in Combination with Oral Methotrexate for KRAS Mutated Non-Small Cell Lung Cancer (NSCLC) |
| NCT03520842 | [105] |
| Genomically-Guided Treatment Trial in Brain Metastases |
| NCT03994796 | [106] |
| Expanded Access to Ulixertinib (BVD-523) in Patients with Advanced MAPK Pathway-Altered Malignancies |
| NCT04566393 | [107] |
| Safety and Efficacy of NEO212 in Patients with Astrocytoma IDH-mutant, Glioblastoma IDH-wildtype or Brain Metastasis |
| NCT06047379 | [108] |
| Study of Efficacy and Safety of Dabrafenib in Combination with Trametinib in Pediatric Patients with BRAF V600 Mutation Positive LGG or Relapsed or Refractory HGG Tumors |
| NCT02684058 | [109] |
| A Study to Determine Safety, Tolerability and Pharmacokinetics of Oral Dabrafenib in Children and Adolescent Subjects |
| NCT01677741 | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza Barbosa, I.; Pilotto Heming, C.; Moura Neto, V.; Aran, V. The Role of RAS in CNS Tumors: A Key Player or an Overlooked Oncogene? Int. J. Mol. Sci. 2025, 26, 4104. https://doi.org/10.3390/ijms26094104
de Souza Barbosa I, Pilotto Heming C, Moura Neto V, Aran V. The Role of RAS in CNS Tumors: A Key Player or an Overlooked Oncogene? International Journal of Molecular Sciences. 2025; 26(9):4104. https://doi.org/10.3390/ijms26094104
Chicago/Turabian Stylede Souza Barbosa, Isabel, Carlos Pilotto Heming, Vivaldo Moura Neto, and Veronica Aran. 2025. "The Role of RAS in CNS Tumors: A Key Player or an Overlooked Oncogene?" International Journal of Molecular Sciences 26, no. 9: 4104. https://doi.org/10.3390/ijms26094104
APA Stylede Souza Barbosa, I., Pilotto Heming, C., Moura Neto, V., & Aran, V. (2025). The Role of RAS in CNS Tumors: A Key Player or an Overlooked Oncogene? International Journal of Molecular Sciences, 26(9), 4104. https://doi.org/10.3390/ijms26094104