
Autoantibody Profiling in Ulcerative Colitis: Identification of Early Immune Signatures and Disease-Associated Antigens for Improved Diagnosis and Monitoring
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Antibody Profiles in Severe UC vs. Controls
2.2. Protein Array Class Comparison Results of Manifested UC vs. Controls
2.3. Antibody Reactivities Correlated with Disease Activity
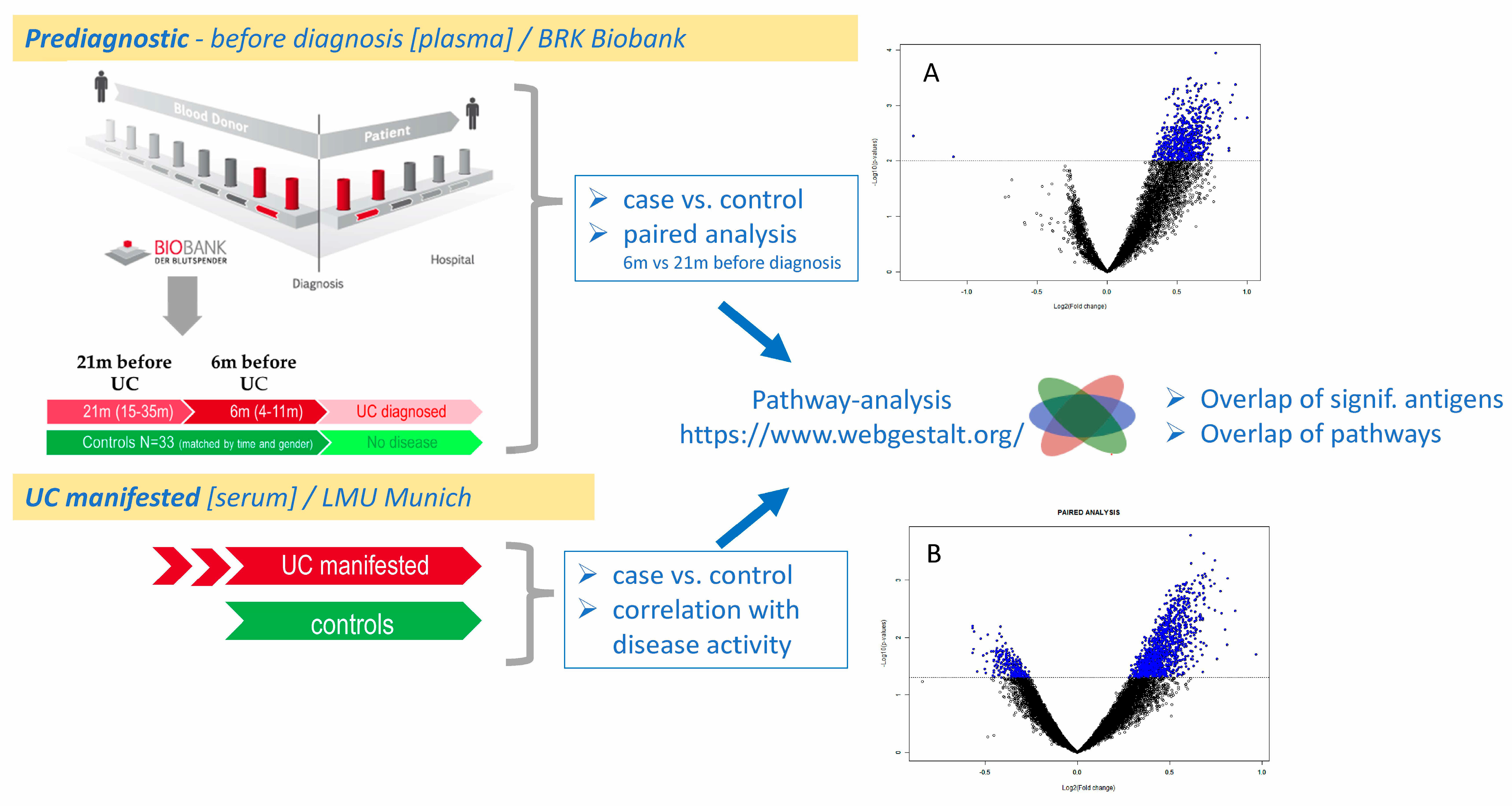
2.4. Antibody Profiles in Pre-Diagnostic UC
2.4.1. Plasma IgG Concentrations in Pre-Diagnostic UC Samples Do Not Increase Until Diagnosis
2.4.2. Differentially Reactive Antigens in Pre-Diagnostic UC
2.4.3. Potential for Early Diagnostics
2.4.4. Comparative Analysis of Autoantibodies in Manifested UC (Serum) vs. Pre-Diagnostic UC (Plasma)
2.4.5. Pathway Analysis of Autoantibodies in Manifested UC (Serum) and Pre-Diagnostic UC (Plasma)
3. Discussion
3.1. Discussion of Top Antigens from Class Comparison and Correlation Analysis with Disease Activity
3.1.1. Top Autoantigens in Manifested UC
3.1.2. Antigens Found Correlated with Clinical Activity Score
3.1.3. Top Autoantigens in Pre-Diagnostic UC
- DCAF5: This protein is involved in the regulation of protein degradation and has been linked to cellular stress responses. Its role in modulating protein stability may influence inflammatory pathways, although specific references to its involvement in UC are limited.
- SRSF9: This gene encodes a splicing factor that plays a role in RNA processing. Alterations in splicing can affect the expression of inflammatory mediators, suggesting a potential link to inflammatory diseases, including UC [11].
- LAMC1: As a component of laminin, LAMC1 is crucial for cell adhesion and maintaining the integrity of the extracellular matrix. Disruption in LAMC1 expression can lead to impaired epithelial barrier function, which is particularly relevant in UC, where barrier integrity is compromised [12].
3.2. Discussion of Selected Pathways
3.2.1. Top 3 Pathways in Manifested Ulcerative Colitis
3.2.2. Pathways Overlapping in Manifested and Pre-Diagnostic UC
3.2.3. Pathways Identified in Pre-Diagnostic UC
3.3. Potential Relevance to UC Treatment Based on the Pathways
Novel Potential Therapeutic Approaches Inspired by Reactome Pathways
3.4. Autoantibodies in UC
4. Materials and Methods
4.1. Samples
4.2. Isolation of Immunoglobulin
4.3. Protein Microarray Processing and Microarray Data Analysis
4.4. Pathway Analysis of Antigens Derived from Protein Microarray Data Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-ASA | 5-aminosalicylic acid |
| A2M | Alpha-2-macroglobulin |
| BRK | Bavarian red cross |
| CD99 | Cluster of differentiation 99 |
| CLTA | Clathrin–AP180 complex |
| DIRAGs | Differentially reactive antigens |
| FC | Fold change |
| IBD | Inflammatory bowel disease |
| IgG | Immunoglobulin G |
| NSG-UC | NOD/ScidIL2Rγnull |
| ORA | Over-representation analysis |
| pANCA | Perinuclear antigens |
| PBMCs | Peripheral blood mononuclear cells |
| PTPN6 | Protein tyrosine phosphatase, non-receptor type 6 |
| RFI | Relative fluorescent intensities |
| SCCAI | Simple clinical colitis activity index |
| TRIM | Tripartite motif |
| TUFM | Tu translation elongation factor mitochondrial |
| UC | Ulcerative colitis |
| WebGestalt | WEB-based Gene SeT AnaLysis Toolkit |
References
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Moura, F.A.; de Andrade, K.Q.; Dos Santos, J.C.F.; Araújo, O.R.P.; Goulart, M.O.F. Antioxidant therapy for treatment of inflammatory bowel disease: Does it work? Redox Biol. 2015, 6, 617–639. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Regev, A.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef]
- Cohavy, O.; Bruckner, D.; Gordon, L.K.; Misra, R.; Wei, B.; Eggena, M.E.; Targan, S.R.; Braun, J. Colonic bacteria express an ulcerative colitis pANCA-related protein epitope. Infect. Immun. 2000, 68, 1542–1548. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Vogl, T.; Segal, E.; Weersma, R.K. Antibody signatures in inflammatory bowel disease: Current developments and future applications. Trends. Mol. Med. 2022, 28, 693–705. [Google Scholar] [CrossRef]
- Mitsuyama, K.; Niwa, M.; Takedatsu, H.; Yamasaki, H.; Kuwaki, K.; Yoshioka, S.; Yamauchi, R.; Fukunaga, S.; Torimura, T. Antibody markers in the diagnosis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1304–1310. [Google Scholar] [CrossRef]
- Jodeleit, H.; Milchram, L.; Soldo, R.; Beikircher, G.; Schönthaler, S.; Al-Amodi, O.; Wolf, E.; Beigel, F.; Weinhäusel, A.; Siebeck, M.; et al. Autoantibodies as diagnostic markers and potential drivers of inflammation in ulcerative colitis. PLoS ONE 2020, 15, e0228615. [Google Scholar] [CrossRef]
- Livanos, A.E.; Dunn, A.; Fischer, J.; Ungaro, R.C.; Turpin, W.; Lee, S.H.; Rui, S.; Riddle, M.S.; Murray, J.A.; Laird, R.M.; et al. Anti-Integrin αvβ6 Autoantibodies Are a Novel Biomarker That Antedate Ulcerative Colitis. Gastroenterology 2023, 164, 619–629. [Google Scholar] [CrossRef]
- Huang, J.H.; Liu, C.Y.; Wu, S.Y.; Chen, W.Y.; Chang, T.H.; Kan, H.W.; Hsieh, S.T.; Ting, J.P.Y.; Wu-Hsieh, B.A. NLRX1 Facilitates Histoplasma capsulatum-Induced LC3-Associated Phagocytosis for Cytokine Production in Macrophages. Front. Immunol. 2018, 9, 2761. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hwang, H.Y.; Ji, E.S.; Kim, J.Y.; Yoo, J.S.; Kwon, H.J. Activation of mitochondrial TUFM ameliorates metabolic dysregulation through coordinating autophagy induction. Commun. Biol. 2021, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Lv, X.; Fang, C.; Liu, C.; Ma, Z.; Liu, K. Silencing TUFM Inhibits Development of Monocrotaline-Induced Pulmonary Hypertension by Regulating Mitochondrial Autophagy via AMPK/mTOR Signal Pathway. Oxidative Med. Cell. Longevity 2022, 1, 4931611. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Oh, H.Y.; Heo, H.S.; Hong, J.E.; Jung, S.J.; Lee, K.W.; Park, J.H.; Hur, C.G.; Park, J.H.Y. Biological features of core networks that result from a high-fat diet in hepatic and pulmonary tissues in mammary tumour-bearing, obesity-resistant mice. Br. J. Nutr. 2013, 110, 241–255. [Google Scholar] [CrossRef]
- Sarabi, A.S.; Shen, H.; Wang, Y.; Hoffer, B.J.; Bäckman, C.M. Gene expression patterns in mouse cortical penumbra after focal ischemic brain injury and reperfusion. J. Neurosci. Res. 2008, 86, 2912–2924. [Google Scholar] [CrossRef]
- Abaturov, A.E.; Nikulina, A.A. Functional annotation of lactase gene and its distal enhancer MCM6 for prediction of metabolically unhealthy obesity. Pediatr. Pol.-Pol. J. Paediatr. 2023, 98, 16–22. [Google Scholar] [CrossRef]
- Sekelova, Z.; Polansky, O.; Stepanova, H.; Fedr, R.; Faldynova, M.; Rychlik, I.; Vlasatikova, L. Different roles of CD4, CD8 and γδ T-lymphocytes in naive and vaccinated chickens during Salmonella Enteritidis infection. Proteomics 2017, 17, 1700073. [Google Scholar] [CrossRef]
- Targan, S.R.; Karp, L.C. Defects in mucosal immunity leading to ulcerative colitis. Immunol. Reviews. 2005, 206, 296–305. [Google Scholar] [CrossRef]
- Ghia, J.; Li, N.; Wang, H.; Collins, M.; Deng, Y.; El–Sharkawy, R.T.; Côté, F.; Mallet, J.; Khan, W.I. Serotonin Has a Key Role in Pathogenesis of Experimental Colitis. Gastroenterology 2009, 137, 1649–1660. [Google Scholar] [CrossRef]
- Li, J.; Pan, X.; Ren, Z.; Li, B.; Liu, H.; Wu, C.; Dong, X.; Vos, P.d.; Pan, L.-L.; Sun, J. Protein arginine methyltransferase 2 (PRMT2) promotes dextran sulfate sodium-induced colitis by inhibiting the SOCS3 promoter via histone H3R8 asymmetric dimethylation. Br. J. Pharmacol. 2022, 179, 141–158. [Google Scholar] [CrossRef]
- Wang, X.; Li, D.; Zhang, Y.; Wu, S.; Tang, F. Costus root granules improve ulcerative colitis through regulation of TGF-β mediation of the PI3K/AKT signaling pathway. Exp. Ther. Med. 2018, 15, 4477–4484. Available online: http://www.spandidos-publications.com/10.3892/etm.2018.5946 (accessed on 24 February 2025). [CrossRef] [PubMed]
- Poddar, D.; Kaur, R.; Baldwin, W.M.; Mazumder, B. L13a-dependent translational control in macrophages limits the pathogenesis of colitis. Cell. Mol. Immunol. 2016, 13, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Yam, A.Y.; Xia, Y.; Lin, H.T.J.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, C.; Saud, S.M.; Lu, X.; Liu, L.; Fang, L.; Zhang, X.; Hu, J.; Li, W. Effect of Kuijie Granule on the Expression of TGF-β/Smads Signaling Pathway in Patients with Ulcerative Colitis. Evid.-Based Complement. Altern. Med. 2016, 1, 2601830. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.T. Current and Emerging Biologics for Ulcerative Colitis. Gut Liver. 2015, 9, 18–27. [Google Scholar] [CrossRef]
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. 2020, 17, 15. [Google Scholar] [CrossRef]
- Guo, M.; Wang, R.; Geng, J.; Li, Z.; Liu, M.; Lu, X.; Wei, J.; Liu, M. Human TFF2-Fc Fusion Protein Alleviates DSS-induced Ulcerative Colitis in C57bl/6 Mice by Promoting Intestinal Epithelial Cells Repair and Inhibiting Macrophage Inflammation. Inflammopharmacology 2023, 31, 1387–1404. [Google Scholar] [CrossRef]
- Zhang, T. Identifying potential drug targets for the treatment of ulcerative colitis using mendelian randomization combined with co-localization analysis. Res. Sq. 2024. [Google Scholar] [CrossRef]
- Huang, T.Q. Bergenin Alleviates Ulcerative Colitis by Decreasing Gut Commensal Bacteroides Vulgatus-Mediated Elevated Branched-Chain Amino Acids. J. Agric. Food Chem. 2024, 72, 3606–3621. [Google Scholar] [CrossRef]
- Xiao, B.; Zhang, Z.; Viennois, É.; Kang, Y.; Zhang, M.; Han, M.K.; Chen, j.; Merlin, D. Combination Therapy for Ulcerative Colitis: Orally Targeted Nanoparticles Prevent Mucosal Damage and Relieve Inflammation. Theranostics 2016, 6, 2250–2266. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent Autophagy via Upstream TLR4-MyD88-MAPK Signalling and Downstream NF-κB Pathway Quenches Intestinal Inflammation and Oxidative Stress Injury. Ebiomedicine 2018, 35, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, M.; Martin, J.C.; Mesin, L.; Livanos, A.E.; Castro-Dopico, T.; Huang, R.; Petralia, F.; Magri, G.; Kumar, S.; Zhao, Q.; et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat. Med. 2022, 28, 766–779. [Google Scholar] [CrossRef]
- He, T.; Wang, K.; Zhao, P.; Zhu, G.; Yin, X.; Zhang, Y.; Zhang, Z.; Zhao, K.; Wang, Z.; Wang, K. Integrative computational approach identifies immune-relevant biomarkers in ulcerative colitis. FEBS Open Bio. 2022, 12, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Giannos, P.; Triantafyllidis, K.K.; Giannos, G.; Kechagias, K.S. SPP1 in infliximab resistant ulcerative colitis and associated colorectal cancer: An analysis of differentially expressed genes. Eur. J. Gastroenterol. Hepatol. 2022, 34, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Ipek, S.; Yalcin, H.; Toprak, B. Association between disease activity and ischaemia-modified albumin in patients with ulcerative colitis. Gastroenterol. Rev./Przegląd Gastroenterol. 2022, 17, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, R.S.; Ayres, R.C.; Pounder, R.E.; Allan, R.N. A simple clinical colitis activity index. Gut 1998, 43, 29–32. [Google Scholar] [CrossRef]
- Milchram, L.; Kulovics, R.; Sonntagbauer, M.; Schönthaler, S.; Vierlinger, K.; Dorfer, C.; Cameron, C.; Saydam, O.; Weinhäusel, A. Antibody Profiling and In Silico Functional Analysis of Differentially Reactive Antibody Signatures of Glioblastomas and Meningiomas. Int. J. Mol. Sci. 2023, 24, 1411. [Google Scholar] [CrossRef]

{kind=link}
| UC n = 49 | Active UC n = 19 | Non-UC n = 23 | |
|---|---|---|---|
| Age (years) | |||
| Mean (SD) | 38.5 (15.6) | 36.13 | 36.7 (15.9) |
| Range | 24–74 | 19–71 | 21–59 |
| Gender (% male) | 46 | 42 | 42 |
| Duration of UC (years) | |||
| Mean (SD) | 11.6 (9.53) | 11.46 (10.97) | |
| Range | 1–40 | 1–39 | |
| SCCAI | |||
| Mean (SD) | 3.04 (2.79) | 6.42 (2.19) | |
| Range | 0–13 | 5–13 | |
| Treatment (current) | |||
| TNFα-blocker | 20 | 6 | |
| Glucocorticoids | 13 | 8 | |
| Mesalazine | 26 | 9 | |
| Immuno-suppressive | 6 | 3 | |
| No | 10 | 3 | |
| Atopic Dermatitis | 3 |
| Parametric p-Value | FDR | Geom Mean of Intensities in Class 1 (UC) | Geom Mean of Intensities in Class 2 (Contr) | Fold Change | UniqueID | Name (Updated) |
|---|---|---|---|---|---|---|
| 0.0016529 | 0.0795 | 1282.1 | 640.82 | 2 | RZPDp9027O0413Q | DMPK |
| 0.0004156 | 0.0795 | 1788.2 | 947.7 | 1.89 | RZPDp9027K1313Q | HES1 |
| 0.001753 | 0.0795 | 1818.41 | 962.46 | 1.89 | RZPDp9027F128Q | GGA1 |
| 0.0006475 | 0.0795 | 1283.67 | 690.63 | 1.86 | RZPDp9027A1813Q | RPS29 |
| 0.0008848 | 0.0795 | 1485.46 | 805.73 | 1.84 | RZPDp9027C1612Q | CD99 |
| 0.005878 | 0.0796 | 937.25 | 513.29 | 1.83 | RZPDp9027B0418Q | TRIM27 |
| 0.0065022 | 0.0825 | 2056.95 | 1124.54 | 1.83 | RZPDp9027B186Q | MCM3AP |
| 0.0013648 | 0.0795 | 2432.38 | 1374.53 | 1.77 | RZPDp9027A1412Q | POGZ |
| 0.0015698 | 0.0795 | 2069.3 | 1180.18 | 1.75 | RZPDp9027H0515Q | SLC16A8 |
| 0.0018793 | 0.0795 | 856.75 | 490.21 | 1.75 | RZPDp9027C1713Q | FLAD1 |
| 0.0003951 | 0.0795 | 354.27 | 203.84 | 1.74 | RZPDp9027D1013Q | NBPF9 |
| 0.001114 | 0.0795 | 467.47 | 267.9 | 1.74 | RZPDp9027J0210Q | SCAP |
| 0.0036918 | 0.0795 | 2389.08 | 1371.05 | 1.74 | RZPDp9027H1111Q | CD99 |
| 0.00383 | 0.0795 | 541.62 | 311.4 | 1.74 | RZPDp9027C138Q | IP6K2 |
| 0.0009492 | 0.0795 | 7994.23 | 4619.48 | 1.73 | RZPDp9027J2111Q | POLR3H |
| 0.0012119 | 0.0795 | 1889.77 | 1089.48 | 1.73 | RZPDp9027D2216Q | RPS17 |
| 0.0023996 | 0.0795 | 810.49 | 469.71 | 1.73 | RZPDp9027K2414Q | UBXN4 |
| 0.0034252 | 0.0795 | 512.57 | 295.68 | 1.73 | RZPDp9027B1813Q | YES1 |
| 0.0008638 | 0.0795 | 1072.27 | 622.45 | 1.72 | RZPDp9027A1312Q | AP2S1 |
| 0.0025636 | 0.0795 | 1071.97 | 624.53 | 1.72 | RZPDp9027L1713Q | SEPTIN7P14 |
| (A) Median Normalised | Correlation Coefficient | Parametric p-Value | Name |
|---|---|---|---|
| 1 | −0.444 | 0.0013822 | PODXL2 |
| 2 | −0.444 | 0.0013919 | FBLL1 |
| 3 | −0.437 | 0.00171 | PHF19 |
| 4 | −0.416 | 0.0029337 | PRPF3 |
| 5 | −0.414 | 0.0031113 | CLTA |
| 6 | −0.413 | 0.0032041 | TMEM44 |
| 7 | −0.406 | 0.0037712 | CHAMP1 |
| 8 | −0.404 | 0.0039976 | TTLL7 |
| 9 | −0.396 | 0.0048621 | CLEC11A |
| 10 | −0.393 | 0.0051929 | PIPSL |
| 11 | −0.391 | 0.0054845 | ATXN10 |
| 12 | −0.391 | 0.0055268 | KNDC1 |
| 13 | −0.39 | 0.00566 | ZWINT |
| 14 | −0.386 | 0.0062099 | TUFM |
| 15 | −0.379 | 0.0071869 | USP34 |
| 16 | −0.378 | 0.0074131 | SUPV3L1 |
| 17 | −0.377 | 0.0076329 | A2M |
| 18 | −0.376 | 0.0078365 | PAICS |
| 19 | 0.374 | 0.0080463 | PDIA4 |
| 20 | −0.373 | 0.0082321 | CCT8 |
| 21 | −0.371 | 0.008659 | XRCC6 |
| 22 | −0.371 | 0.0087122 | ROBO3 |
| 23 | −0.368 | 0.0092002 | OLA1 |
| 24 | −0.368 | 0.0093 | BSG |
| 25 | −0.367 | 0.0094562 | PRMT2 |
| 26 | −0.365 | 0.0099094 | MEIS3 |
| (B) Un- Normalised | Correlation Coefficient | Parametric p-Value | Name |
| 1 | −0.57 | 5.40 × 10−5 | DCAF13 |
| 2 | −0.53 | 9.01 × 10−5 | ACO2 |
| 3 | −0.528 | 9.62 × 10−5 | C17orf70 |
| 4 | −0.524 | 0.0001106 | TPP2 |
| 5 | −0.522 | 0.0001681 | RPL7A |
| 6 | −0.51 | 0.0001831 | VARS2 |
| 7 | −0.506 | 0.0002098 | RAB5C |
| 8 | −0.502 | 0.0002377 | DHX30 |
| 9 | −0.5 | 0.0002513 | PTPN6 |
| 10 | −0.487 | 0.0003836 | MINA |
| 11 | −0.477 | 0.0005248 | CNTD1 |
| 12 | −0.476 | 0.0005513 | MCM6 |
| 13 | −0.475 | 0.0005583 | ZWINT |
| 14 | −0.472 | 0.0006137 | RSRP1 |
| 15 | −0.472 | 0.0006261 | GOLGA2 |
| 16 | −0.471 | 0.0006396 | CLTA |
| 17 | −0.464 | 0.0007813 | TUFM |
| 18 | −0.464 | 0.0007823 | CCDC94 |
| 19 | −0.46 | 0.0008893 | HSF1 |
| 20 | −0.458 | 0.0009435 | CCT2 |
| 21 | −0.456 | 0.0009975 | FLNA |
| UC Individuals | Year of Diagnosis | Age at Diagnosis (y) | Gender | Blood Group (AB0) | Time (month) Sample Taken Prior Diagnosis | IgG Concentration * [mg/mL plasma] | IgG Increase (%) ** | Delta T3–Tx (m) ** | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | IgG T1 | IgG T2 | IgG T3 | |||||||
| case 1 | 2008 | 45 | M | 0 | 22 | 20 | 4 | 7.95 | 8.70 | 9.24 | 6% | 16 |
| case 2 *** | 2004 | 35 | W | A | 19 | 16 | 4 | 7.17 | 7.47 | 8.78 | 22% | 15 |
| case 3 *** | 2005 | 61 | M | 0 | 15 | 10 | 6 | 7.01 | 6.37 | 6.59 | –6% | 9 |
| case 4 | 2004 | 45 | M | A | 7 | 4 | 1 | 7.32 | 7.59 | 8.34 | ||
| case 5 | 2005 | 46 | W | A | 17 | 12 | 4 | 9.64 | 8.94 | 7.11 | –26% | 13 |
| case 6 | 2011 | 35 | M | 0 | 55 | 53 | 50 | 7.07 | 7.49 | 7.31 | ||
| case 7 | 2009 | 42 | W | A | 34 | 24 | 11 | 7.17 | 5.33 | 8.18 | 53% | 13 |
| case 8 | 2009 | 31 | W | A | 25 | 22 | 10 | 8.29 | 5.87 | 6.27 | 7% | 12 |
| case 9 | 2010 | 43 | M | 0 | 65 | 22 | 3 | 8.71 | 5.91 | 7.60 | 29% | 19 |
| case 10 | 2012 | 49 | W | B | 21 | 14 | 11 | 10.70 | 5.23 | 4.33 | –60% | 10 |
| case 11 | 2012 | 44 | M | 0 | 35 | 20 | 11 | 5.47 | 5.48 | 7.03 | 29% | 24 |
| median | 44 | 6 | 7.31 | 6.8% | 13 | |||||||
| Protein/Gene Symbol | Parametric p-Value | Fold Change (T3 vs. T-Early) |
|---|---|---|
| DCAF5 | 0.0034474 | 1.81 |
| SRSF9 | 0.0009296 | 1.76 |
| LAMC1 | 0.0131338 | 1.75 |
| SULT1A3 | 0.0072182 | 1.74 |
| EVL | 0.0038099 | 1.72 |
| CKAP5 | 0.0015792 | 1.71 |
| TINF2 | 0.0235447 | 1.69 |
| DISP3 | 0.0004463 | 1.68 |
| NAP1L4 | 0.0033596 | 1.68 |
| PTPRE | 0.001167 | 1.67 |
| EIF2S3 | 0.0006555 | 1.66 |
| TIAL1 | 0.0012767 | 1.65 |
| FAM13A | 0.0010115 | 1.64 |
| USP11 | 0.0030261 | 1.64 |
| FMNL2 | 0.0008524 | 1.62 |
| HNRNPA2B1 | 0.0139107 | 1.62 |
| PPID | 0.0003352 | 1.61 |
| MAGED2 | 0.0007698 | 1.61 |
| SUGP2 | 0.001397 | 1.61 |
| FAU | 0.0020518 | 1.61 |
| ANTIGENS | Parametric p-Value | t-Value | Fold Change |
|---|---|---|---|
| DCAF5 | 0.0034474 | 3.397 | 1.81 |
| PPID | 0.0003352 | 4.476 | 1.61 |
| GBAS | 0.0008214 | 4.059 | 1.6 |
| LRP5 | 0.001681 | 3.728 | 1.58 |
| PKM | 0.000819 | 4.06 | 1.57 |
| WNK2 | 0.001018 | 3.96 | 1.55 |
| POGLUT1 | 0.0011713 | 3.895 | 1.54 |
| AAAS | 0.0001634 | 4.815 | 1.53 |
| LMO4 | 0.0025234 | 3.541 | 1.52 |
| AP2S1 | 0.0015832 | 3.756 | 1.51 |
| SLC22A17 | 0.0030934 | 3.447 | 1.5 |
| ANTIGENS | Parametric p-Value | t-Value | Fold Change |
|---|---|---|---|
| EVL | 0.0002057 | 4.818 | 2.1 |
| HNRNPA2B1 | 0.0000997 | 5.184 | 2.1 |
| CD58 | 0.0000735 | 5.34 | 2.06 |
| EEF1A1 | 0.0004424 | 4.439 | 2.01 |
| DRG1 | 0.0000945 | 5.211 | 1.98 |
| DHX8 | 0.0001773 | 4.893 | 1.95 |
| GNAI2 | 0.0001624 | 4.936 | 1.95 |
| ALKBH2 | 0.0001302 | 5.048 | 1.95 |
| BCAR1 | 0.0000853 | 5.264 | 1.93 |
| TIAL1 | 0.0001786 | 4.889 | 1.92 |
| FADS1 | 0.0000979 | 5.193 | 1.92 |
| EIF2S3 | 0.0001117 | 5.126 | 1.91 |
| PTCHD2 | 0.0000894 | 5.24 | 1.91 |
| LAMTOR1 | 0.000414 | 4.471 | 1.89 |
| RNF10 | 0.0003557 | 4.546 | 1.89 |
| FAM209B | 0.0003529 | 4.55 | 1.89 |
| CDCA4 | 0.0002964 | 4.636 | 1.89 |
| FAM13A | 0.000187 | 4.866 | 1.89 |
| SUGP2 | 0.0000849 | 5.266 | 1.89 |
| CPNE1 | 0.0003801 | 4.513 | 1.88 |
| VWF | 0.0003278 | 4.586 | 1.88 |
| GOT1 | 0.0000907 | 5.232 | 1.88 |
| ZNF84 | 0.0002809 | 4.663 | 1.87 |
| RUNDC3A | 0.0001665 | 4.924 | 1.87 |
| FMNL2 | 0.0001064 | 5.151 | 1.87 |
| RNASEK | 0.0001831 | 4.876 | 1.86 |
| C17orf62 | 0.0001416 | 5.006 | 1.84 |
| FAM131A | 0.0004892 | 4.389 | 1.83 |
| MINK1 | 0.000285 | 4.656 | 1.83 |
| SH3BGRL3 | 0.0001401 | 5.011 | 1.83 |
| SIRT6 | 0.0002133 | 4.8 | 1.81 |
| CKB | 0.0002853 | 4.655 | 1.8 |
| PLXNB2 | 0.0004156 | 4.47 | 1.78 |
| CCT3 | 0.0003019 | 4.627 | 1.78 |
| UBE2L3 | 0.0002807 | 4.663 | 1.78 |
| USP28 | 0.0001105 | 5.132 | 1.77 |
| TTC19 | 0.0001416 | 5.006 | 1.76 |
| EIF4A2 | 0.0004477 | 4.433 | 1.75 |
| FTSJD2 | 0.0003739 | 4.522 | 1.75 |
| R3HDM1 | 0.0004045 | 4.483 | 1.74 |
| PPID | 0.0004417 | 4.44 | 1.73 |
| WNK2 | 0.000428 | 4.455 | 1.73 |
| HSP90AB1 | 0.0001805 | 4.883 | 1.73 |
| PDHA1 | 0.0004512 | 4.429 | 1.72 |
| KIAA1731 | 0.0003517 | 4.552 | 1.72 |
| RPL36A | 0.0002 | 4.832 | 1.72 |
| DST | 0.0001255 | 5.067 | 1.71 |
| CAPZA2 | 0.0001205 | 5.087 | 1.71 |
| EIF3G | 0.0004173 | 4.468 | 1.7 |
| SRSF2 | 0.0003381 | 4.571 | 1.7 |
| DYNC1I2 | 0.0003514 | 4.552 | 1.68 |
| NARS2 | 0.0002444 | 4.732 | 1.67 |
| ZCCHC11 | 0.0004672 | 4.412 | 1.65 |
| GTF2I | 0.0004434 | 4.438 | 1.65 |
| MSL1 | 0.0004614 | 4.418 | 1.61 |
| AAAS | 0.0004851 | 4.394 | 1.6 |
| (A) | Gene Set | Description | Size | Expect | Ratio | p Value | FDR |
|---|---|---|---|---|---|---|---|
| 1 | R-HSA-5653656 | Vesicle-mediated transport | 350 | 38.576 | 1.7109 | 3.6111 × 10−6 | 0.0045419 |
| 2 | R-HSA-199991 | Membrane Trafficking | 327 | 36.041 | 1.7203 | 6.1879 × 10−6 | 0.0045419 |
| 3 | R-HSA-446203 | Asparagine N-linked glycosylation | 134 | 14.769 | 2.1667 | 0.000012865 | 0.0062954 |
| 4 | R-HSA-2132295 | MHC class II antigen presentation | 75 | 8.2662 | 2.5405 | 0.000034295 | 0.0096775 |
| 5 | R-HSA-6807878 | COPI-mediated anterograde transport | 59 | 6.5028 | 2.7681 | 0.00003565 | 0.0096775 |
| 6 | R-HSA-389960 | Formation of tubulin folding intermediates by CCT/TriC | 18 | 1.9839 | 4.5365 | 0.000043264 | 0.0096775 |
| 7 | R-HSA-199977 | ER to Golgi Anterograde Transport | 82 | 9.0377 | 2.4342 | 0.000046146 | 0.0096775 |
| 8 | R-HSA-389957 | Prefoldin mediated transfer of substrate to CCT/TriC | 23 | 2.535 | 3.9448 | 0.000072494 | 0.013303 |
| 9 | R-HSA-390450 | Folding of actin by CCT/TriC | 9 | 0.99195 | 6.0487 | 0.00010891 | 0.014974 |
| 10 | R-HSA-437239 | Recycling pathway of L1 | 33 | 3.6371 | 3.2993 | 0.00011214 | 0.014974 |
| 11 | R-HSA-389958 | Cooperation of Prefoldin and TriC/CCT in actin and tubulin folding | 24 | 2.6452 | 3.7804 | 0.0001122 | 0.014974 |
| 12 | R-HSA-390466 | Chaperonin-mediated protein folding | 44 | 4.8495 | 2.8869 | 0.00016066 | 0.018747 |
| 13 | R-HSA-9646399 | Aggrephagy | 25 | 2.7554 | 3.6292 | 0.00016885 | 0.018747 |
| 14 | R-HSA-8866427 | VLDLR internalisation and degradation | 13 | 1.4328 | 4.8855 | 0.00017879 | 0.018747 |
| 15 | R-HSA-948021 | Transport to the Golgi and subsequent modification | 90 | 9.9195 | 2.2179 | 0.00020769 | 0.020326 |
| 16 | R-HSA-5663205 | Infectious disease | 527 | 58.084 | 1.429 | 0.00023271 | 0.021332 |
| 17 | R-HSA-177504 | Retrograde neurotrophin signalling | 10 | 1.1022 | 5.4438 | 0.00024703 | 0.021332 |
| 18 | R-HSA-390471 | Association of TriC/CCT with target proteins during biosynthesis | 22 | 2.4248 | 3.7117 | 0.00029508 | 0.024066 |
| 19 | R-HSA-901042 | Calnexin/calreticulin cycle | 14 | 1.543 | 4.5365 | 0.00032389 | 0.02411 |
| 20 | R-HSA-9734009 | Defective Intrinsic Pathway for Apoptosis | 18 | 1.9839 | 4.0325 | 0.00032847 | 0.02411 |
| 21 | R-HSA-391251 | Protein folding | 48 | 5.2904 | 2.6463 | 0.00044476 | 0.031091 |
| 22 | R-HSA-9663891 | Selective autophagy | 44 | 4.8495 | 2.6807 | 0.0006185 | 0.041271 |
| 23 | R-HSA-373760 | L1CAM interactions | 61 | 6.7232 | 2.3798 | 0.00066087 | 0.042181 |
| 24 | R-HSA-9824446 | Viral Infection Pathways | 443 | 48.826 | 1.4337 | 0.00071004 | 0.043431 |
| (B) | Gene Set | Description | Size | Expect | Ratio | p Value | FDR |
| 1 | R-HSA-156842 | Eukaryotic Translation Elongation | 81 | 16.612 | 2.1069 | 2.4876 × 10−6 | 0.001445 |
| 2 | R-HSA-156827 | L13a-mediated translational silencing of Ceruloplasmin expression | 99 | 20.303 | 1.9701 | 3.7689 × 10−6 | 0.001445 |
| 3 | R-HSA-72706 | GTP hydrolysis and joining of the 60S ribosomal subunit | 99 | 20.303 | 1.9701 | 3.7689 × 10−6 | 0.001445 |
| 4 | R-HSA-72689 | Formation of a pool of free 40S subunits | 89 | 18.252 | 2.0271 | 3.9373 × 10−6 | 0.001445 |
| 5 | R-HSA-72613 | Eukaryotic Translation Initiation | 104 | 21.329 | 1.9223 | 5.9519 × 10−6 | 0.0014562 |
| 6 | R-HSA-72737 | Cap-dependent Translation Initiation | 104 | 21.329 | 1.9223 | 5.9519 × 10−6 | 0.0014562 |
| 7 | R-HSA-2262752 | Cellular responses to stress | 445 | 91.262 | 1.3916 | 0.00001093 | 0.0022921 |
| 8 | R-HSA-8953897 | Cellular responses to stimuli | 447 | 91.672 | 1.3854 | 0.000013969 | 0.0025634 |
| 9 | R-HSA-156902 | Peptide chain elongation | 77 | 15.791 | 2.0264 | 0.000017954 | 0.0029286 |
| 10 | R-HSA-192823 | Viral mRNA Translation | 75 | 15.381 | 2.0154 | 0.000027611 | 0.0040532 |
| 11 | R-HSA-3371497 | HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand | 38 | 7.7932 | 2.438 | 0.000045552 | 0.0059558 |
| 12 | R-HSA-72764 | Eukaryotic Translation Termination | 77 | 15.791 | 1.9631 | 0.000050981 | 0.0059558 |
| 13 | R-HSA-9633012 | Response of EIF2AK4 (GCN2) to amino acid deficiency | 84 | 17.227 | 1.9156 | 0.000052742 | 0.0059558 |
| 14 | R-HSA-975956 | Nonsense Mediated Decay (NMD) independent of the Exon Junction Complex (EJC) | 81 | 16.612 | 1.9264 | 0.000060108 | 0.0063027 |
| 15 | R-HSA-2408522 | Selenoamino acid metabolism | 93 | 19.073 | 1.8351 | 0.000089636 | 0.0083464 |
| 16 | R-HSA-2408557 | Selenocysteine synthesis | 79 | 16.202 | 1.9134 | 0.000090969 | 0.0083464 |
| 17 | R-HSA-71291 | Metabolism of amino acids and derivatives | 199 | 40.812 | 1.5437 | 0.000096924 | 0.0083697 |
| 18 | R-HSA-9711097 | Cellular response to starvation | 109 | 22.354 | 1.7447 | 0.00013029 | 0.010626 |
| 19 | R-HSA-168273 | Influenza Viral RNA Transcription and Replication | 110 | 22.559 | 1.7288 | 0.00016304 | 0.011661 |
| 20 | R-HSA-927802 | Nonsense-Mediated Decay (NMD) | 92 | 18.868 | 1.802 | 0.00017081 | 0.011661 |
| 21 | R-HSA-975957 | Nonsense Mediated Decay (NMD) enhanced by the Exon Junction Complex (EJC) | 92 | 18.868 | 1.802 | 0.00017081 | 0.011661 |
| 22 | R-HSA-72702 | Ribosomal scanning and start codon recognition | 54 | 11.074 | 2.0768 | 0.00017476 | 0.011661 |
| 23 | R-HSA-72695 | Formation of the ternary complex, and subsequently, the 43S complex | 48 | 9.844 | 2.1333 | 0.00021176 | 0.013516 |
| 24 | R-HSA-72649 | Translation initiation complex formation | 55 | 11.28 | 2.0391 | 0.00024325 | 0.014879 |
| 25 | R-HSA-5663205 | Infectious disease | 527 | 108.08 | 1.2861 | 0.00029014 | 0.016485 |
| 26 | R-HSA-1799339 | SRP-dependent cotranslational protein targeting to membrane | 87 | 17.842 | 1.7935 | 0.00029196 | 0.016485 |
| 27 | R-HSA-72662 | Activation of the mRNA upon binding of the cap-binding complex and eIFs, and subsequent binding to 43S | 56 | 11.485 | 2.0027 | 0.00033432 | 0.018177 |
| 28 | R-HSA-8876725 | Protein methylation | 9 | 1.8457 | 3.7925 | 0.00036366 | 0.019066 |
| 29 | R-HSA-9612973 | Autophagy | 85 | 17.432 | 1.7783 | 0.00043017 | 0.020496 |
| 30 | R-HSA-168255 | Influenza Infection | 126 | 25.84 | 1.6254 | 0.00043201 | 0.020496 |
| 31 | R-HSA-422475 | Axon guidance | 316 | 64.806 | 1.3733 | 0.00043281 | 0.020496 |
| 32 | R-HSA-9646399 | Aggrephagy | 25 | 5.1271 | 2.5356 | 0.00045665 | 0.020949 |
| 33 | R-HSA-72766 | Translation | 195 | 39.991 | 1.4753 | 0.00061669 | 0.027433 |
| 34 | R-HSA-9675108 | Nervous system development | 328 | 67.267 | 1.3528 | 0.00064718 | 0.027943 |
| 35 | R-HSA-9010553 | Regulation of expression of SLITs and ROBOs | 133 | 27.276 | 1.5765 | 0.00076776 | 0.032131 |
| 36 | R-HSA-8953854 | Metabolism of RNA | 481 | 98.645 | 1.2773 | 0.00078795 | 0.032131 |
| 37 | R-HSA-9824446 | Viral Infection Pathways | 443 | 90.852 | 1.2878 | 0.00088965 | 0.035298 |
| 38 | R-HSA-9735869 | SARS-CoV-1 modulates host translation machinery | 33 | 6.7677 | 2.2164 | 0.0010492 | 0.040532 |
| 39 | R-HSA-389960 | Formation of tubulin folding intermediates by CCT/TriC | 18 | 3.6915 | 2.7089 | 0.001089 | 0.04099 |
| 40 | R-HSA-389958 | Cooperation of Prefoldin and TriC/CCT in actin and tubulin folding | 24 | 4.922 | 2.438 | 0.0011914 | 0.04372 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinhaeusel, A.; Huber, J.; Schoenthaler, S.; Beigel, F.; Noehammer, C.; Vierlinger, K.; Siebeck, M.; Gropp, R. Autoantibody Profiling in Ulcerative Colitis: Identification of Early Immune Signatures and Disease-Associated Antigens for Improved Diagnosis and Monitoring. Int. J. Mol. Sci. 2025, 26, 4086. https://doi.org/10.3390/ijms26094086
Weinhaeusel A, Huber J, Schoenthaler S, Beigel F, Noehammer C, Vierlinger K, Siebeck M, Gropp R. Autoantibody Profiling in Ulcerative Colitis: Identification of Early Immune Signatures and Disease-Associated Antigens for Improved Diagnosis and Monitoring. International Journal of Molecular Sciences. 2025; 26(9):4086. https://doi.org/10.3390/ijms26094086
Chicago/Turabian StyleWeinhaeusel, Andreas, Jasmin Huber, Silvia Schoenthaler, Florian Beigel, Christa Noehammer, Klemens Vierlinger, Matthias Siebeck, and Roswitha Gropp. 2025. "Autoantibody Profiling in Ulcerative Colitis: Identification of Early Immune Signatures and Disease-Associated Antigens for Improved Diagnosis and Monitoring" International Journal of Molecular Sciences 26, no. 9: 4086. https://doi.org/10.3390/ijms26094086
APA StyleWeinhaeusel, A., Huber, J., Schoenthaler, S., Beigel, F., Noehammer, C., Vierlinger, K., Siebeck, M., & Gropp, R. (2025). Autoantibody Profiling in Ulcerative Colitis: Identification of Early Immune Signatures and Disease-Associated Antigens for Improved Diagnosis and Monitoring. International Journal of Molecular Sciences, 26(9), 4086. https://doi.org/10.3390/ijms26094086