
Profiling of Protein-Coding Missense Mutations in Mendelian Rare Diseases: Clues from Structural Bioinformatics

 , , ,
, , ,  and
and
Abstract

1. Introduction
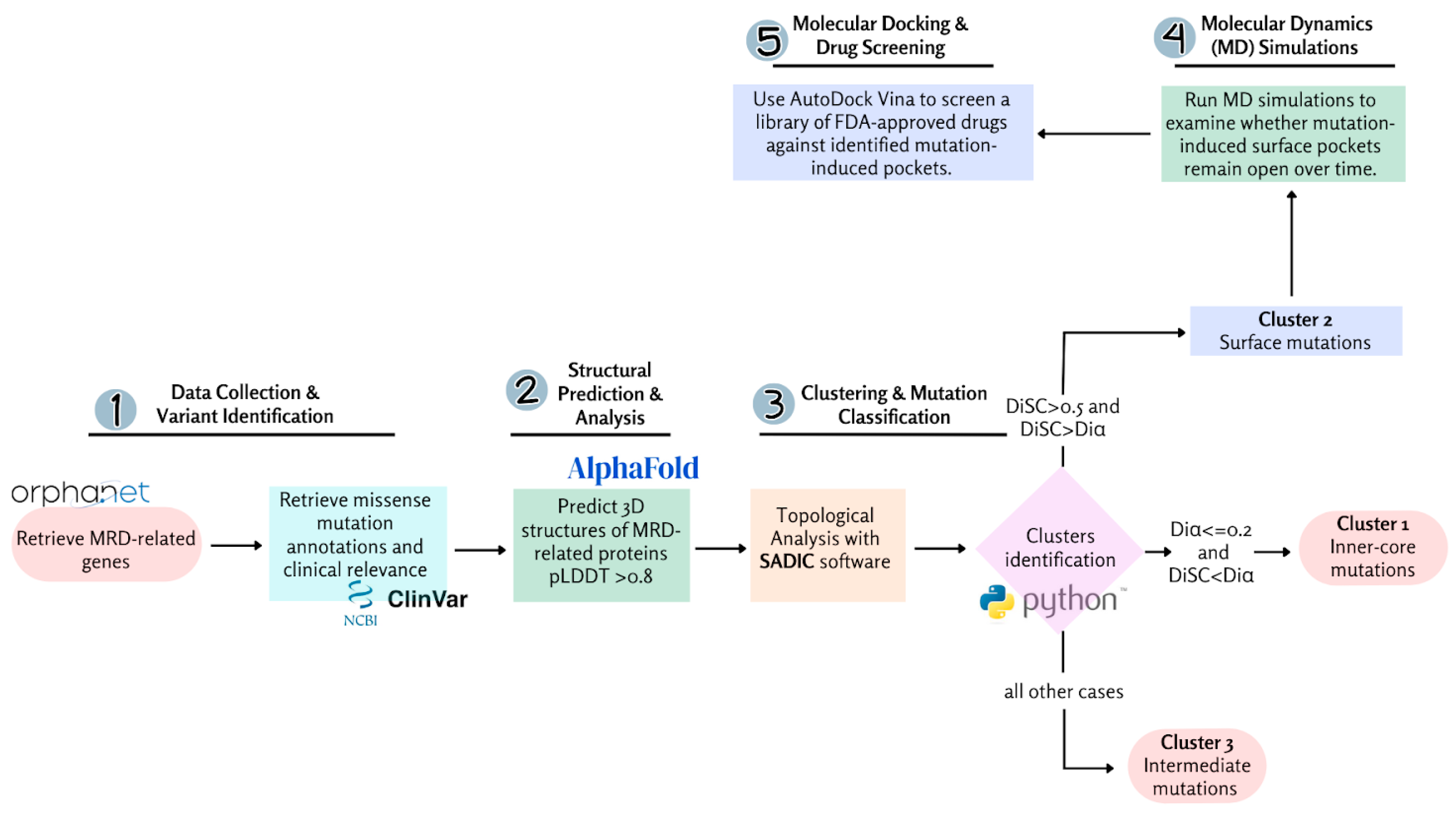
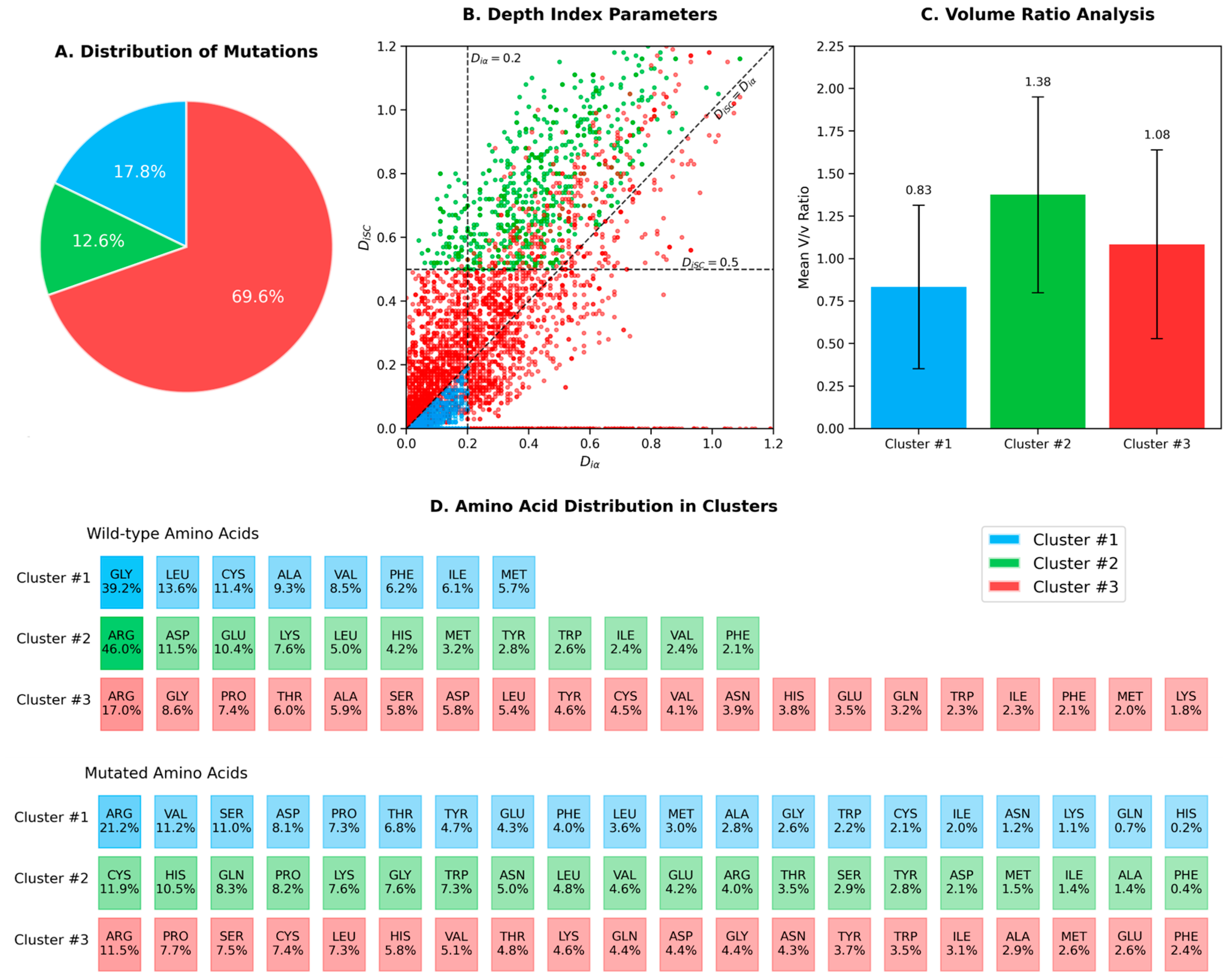
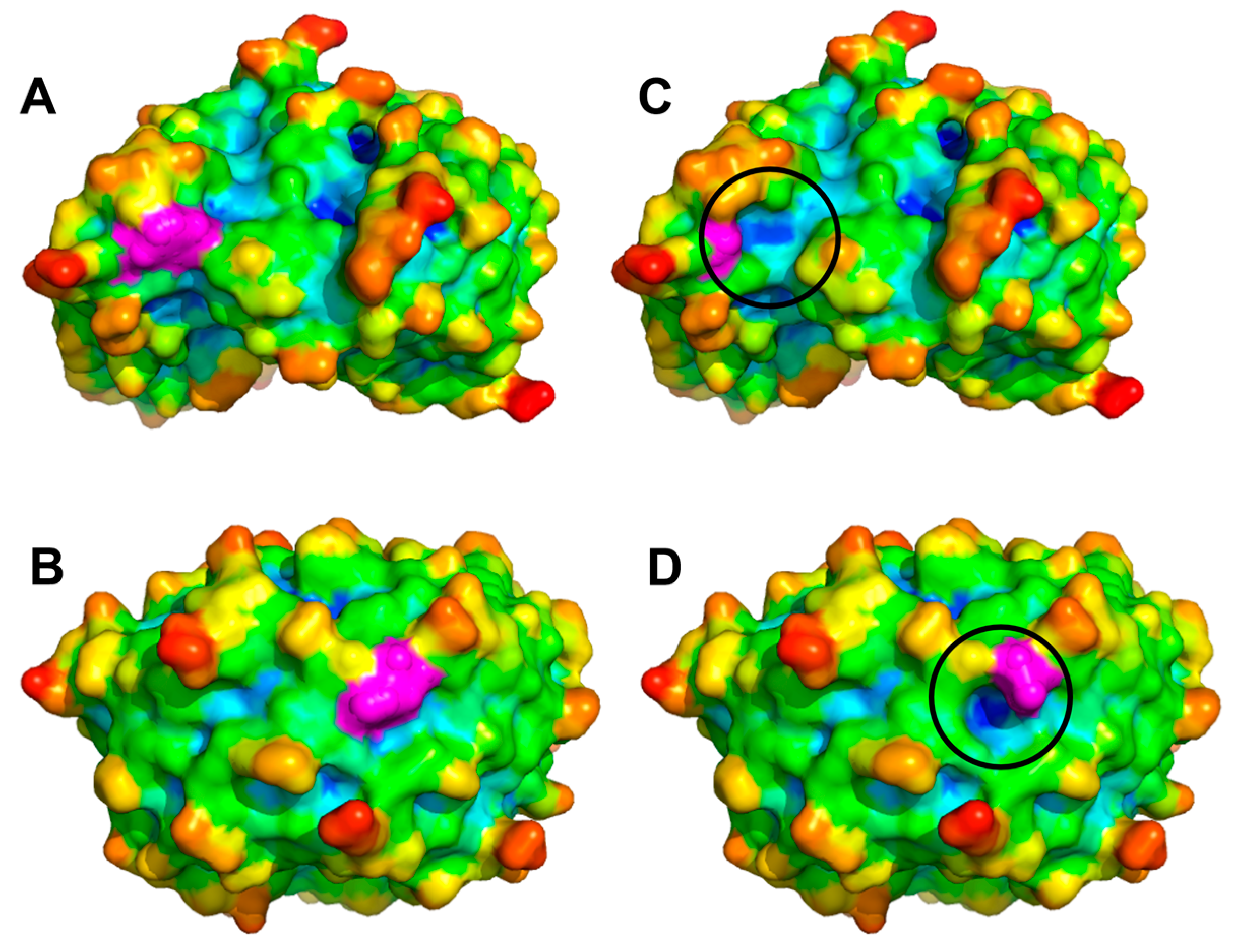
2. Results
3. Discussion
4. Materials and Methods
4.1. Development of the Missense Variant Dataset
4.2. Structure Prediction and Analysis
4.3. Solvent Accessibility Profiling and Burial Analysis
4.4. Molecular Dynamics Simulations
4.5. Molecular Docking Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Rare Diseases; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Vickers, P.J. Challenges and Opportunities in the Treatment of Rare Diseases. Drug Discov. World 2013, 14, 9–16. [Google Scholar]
- Navarrete-Opazo, A.A.; Singh, M.; Tisdale, A.; Cutillo, C.M.; Garrison, S.R. Can you hear us now? The impact of health-care utilization by rare disease patients in the United States. Anesth. Analg. 2021, 23, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R. The burden of rare diseases. Am. J. Med. Genet. A 2019, 179, 885–892. [Google Scholar] [CrossRef]
- List of Rare Disease Organisations. Available online: https://en.wikipedia.org/wiki/List_of_rare_disease_organisations (accessed on 17 September 2024).
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of rare diseases in health information systems: The Orphanet approach to serve a wide range of end users. Hum. Mutat. 2012, 33, 803–808. [Google Scholar] [CrossRef]
- Schieppati, A.; Henter, J.I.; Daina, E.; Aperia, A. Why rare diseases are an important medical and social issue. Lancet 2008, 371, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.P.; Cutillo, C.M.; Lau, L.P.L.; Jonker, A.H.; Rath, A.; Julkowska, D.; Thomson, D.; Terry, S.F.; de Montleau, B.; Ardigò, D.; et al. Future of rare diseases research 2017–2027: An IRDiRC perspective. Clin. Transl. Sci. 2018, 11, 21–27. [Google Scholar] [CrossRef]
- Pavan, S.; Rommel, K.; Mateo Marquina, M.E.; Höhn, S.; Lanneau, V.; Rath, A. Clinical practice guidelines for rare diseases: The Orphanet database. PLoS ONE 2017, 12, e0170365. [Google Scholar] [CrossRef]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Posey, J.E.; O’Donnell-Luria, A.H.; Chong, J.X.; Harel, T.; Jhangiani, S.N.; Coban Akdemir, Z.H.; Buyske, S.; Pehlivan, D.; Carvalho, C.M.B.; Baxter, S.; et al. Insights into genetics, human biology, and disease gleaned from family-based genomic studies. Genet. Med. 2019, 21, 798–812. [Google Scholar] [CrossRef]
- Pradhan, A.; Kalin, T.V.; Kalinichenko, V.V. Genome editing for rare diseases. Curr. Stem Cell Rep. 2020, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tambuyzer, E.; Vandendriessche, B.; Austin, C.P.; Brooks, P.J.; Larsson, K.; Miller Needleman, K.I.; Valentine, J.; Davies, K.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapeutic modalities, progress, and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic modifiers and rare Mendelian disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef]
- Bauer, M.R.; Krämer, A.; Settanni, G.; Jones, R.N.; Ni, X.; Khan Tareque, R.; Fersht, A.R.; Spencer, J.; Joerger, A.C. Targeting cavity-creating p53 cancer mutations with small-molecule stabilizers: The Y220X paradigm. ACS Chem. Biol. 2020, 15, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 pathway: Origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Small-molecule structure correctors target abnormal protein structure and function: Structure corrector rescue of apolipoprotein E4-associated neuropathology. J. Med. Chem. 2012, 55, 8997–9008. [Google Scholar] [CrossRef]
- Alocci, D.; Bernini, A.; Niccolai, N. Atom depth analysis delineates mechanisms of protein intermolecular interactions. Biochem. Biophys. Res. Commun. 2013, 436, 725–729. [Google Scholar] [CrossRef]
- Ehre, C.; Lopes-Pacheco, M.; Laselva, O. Editorial: Mechanisms of action of small molecules on CFTR mutants and the impact on cystic fibrosis pathogenesis. Front. Mol. Biosci. 2024, 11, 1446875. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Visibelli, A.; Finetti, R.; Niccolai, N.; Spiga, O.; Santucci, A. Molecular origins of the Mendelian rare diseases reviewed by Orpha.net: A structural bioinformatics investigation. Int. J. Mol. Sci. 2024, 25, 6953. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ba, A.-N.N.; Ran, X.; Shapovalov, M.V.; Tao, L.; Vakser, I.A.; Dou, Y.; Sun, M.; Katsonis, P.; Lichtarge, O. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Visibelli, A.; Roncaglia, B.; Spiga, O.; Santucci, A. The Impact of Artificial Intelligence in the Odyssey of Rare Diseases. Biomedicines 2023, 11, 887. [Google Scholar] [CrossRef]
- Trezza, A.; Visibelli, A.; Roncaglia, B.; Spiga, O.; Santucci, A. Unsupervised Learning in Precision Medicine: Unlocking Personalized Healthcare through AI. Appl. Sci. 2024, 14, 9305. [Google Scholar] [CrossRef]
- Zhou, L.; Wong, L.; Goh, W.W.B. Understanding missing proteins: A functional perspective. Drug Discov. Today 2018, 23, 644–651. [Google Scholar] [CrossRef]
- Vavilis, T.; Stamoula, E.; Ainatzoglou, A.; Sachinidis, A.; Lamprinou, M.; Dardalas, I.; Vizirianakis, I.S. mRNA in the context of protein replacement therapy. Pharmaceutics 2023, 15, 166. [Google Scholar] [CrossRef]
- Pedrolli, D.B.; Ribeiro, N.V.; Squizato, P.N.; de Jesus, V.N.; Cozetto, D.A.; Team AQA Unesp at iGEM 2017. Engineering microbial living therapeutics: The synthetic biology toolbox. Trends Biotechnol 2019, 37, 100–115. [Google Scholar] [CrossRef]
- Zamyatnin, A.A. Amino acid, peptide, and protein volume in solution. Annu. Rev. Biophys. Bioeng. 1984, 13, 145–165. [Google Scholar] [CrossRef]
- Giess, R.; Holtmann, B.; Braga, M.; Grimm, T.; Müller-Myhsok, B.; Toyka, K.V.; Sendtner, M. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: Potential impact of CNTF as a candidate modifier gene. Am. J. Hum. Genet. 2002, 70, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Ward, M.; Borowsky, J.; Kshirsagar, M.; Lotthammer, J.M.; Oviedo, F.; Ferres, J.L.; Bowman, G.R. Predicting locations of cryptic pockets from single protein structures using the PocketMiner graph neural network. Nat. Commun. 2023, 14, 1177. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Bhakat, S.; Solieva, S.; Bowman, G.R. Accelerating cryptic pocket discovery using AlphaFold. J. Chem. Theory Comput. 2023, 19, 4355–4363. [Google Scholar] [CrossRef] [PubMed]
- Vats, S.; Bobrovs, R.; Söderhjelm, P.; Bhakat, S. AlphaFold-SFA: Accelerated sampling of cryptic pocket opening, protein-ligand binding, and allostery by AlphaFold, slow feature analysis and metadynamics. PLoS ONE 2024, 19, e0307226. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Marziali, S.; Nunziati, G.; Prete, A.L.; Niccolai, N.; Bianchini, M. SADIC v2: A modern implementation of the simple atom depth index calculator. SoftwareX 2024, 27, 101803. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 17 September 2024).
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Janson, G.; Paiardini, A. PyMod 3: A complete suite for structural bioinformatics in PyMOL. Bioinformatics 2021, 37, 1471–1472. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cuong, N.M.; Son, N.T.; Nhan, N.T.; Khanh, P.N.; Huong, T.T.; Tram, N.T.T.; Sgaragli, G.; Ahmed, A.; Trezza, A.; Spiga, O.; et al. Vasorelaxing Activity of R-(-)-3′-Hydroxy-2,4,5-trimethoxydalbergiquinol from Dalbergia tonkinensis: Involvement of Smooth Muscle CaV1.2 Channels. Planta Med. 2020, 86, 284–293. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Side Chain Orientation | Amino Acids | Occurrences |
|---|---|---|---|
| 1 | Diα ≤ 0.2 and DiSC < Diα | Ala, Cys, Gly, Ile, Leu, Met, Phe, Val | 1018 |
| 2 | DiSC > 0.5 and DiSC > Diα | Tyr, Phe, Leu, Ile, Val, Trp, Met Asp, Glu, His, Lys, Arg | 722 |
| 3 | All other cases | all amino acids | 3988 |
| Gene a | PDB b | Aaa N Bbb c | DiSC/Diα d | V/ve |
|---|---|---|---|---|
| SOD1 | 8GSQ | Val149Gly | 1.46 | 2.37 |
| SOD1 | 8GSQ | Leu127Ser | 1.43 | 1.90 |
| SOD1 | 8GSQ | Ile114Thr | 1.23 | 1.45 |
| SOD1 | 8GSQ | Ile152Thr | 1.30 | 1.45 |
| QDPR | 1HDR | Trp108Gly | 3.77 | 3.89 |
| HOGA1 | 3S5N | Trp262Gly | 1.37 | 3.89 |
| TPK1 | 3S4Y | Phe132Ser | 1.54 | 2.17 |
| PAFAH1B1 | 7MT1 | Phe142Ser | 1.24 | 2.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visibelli, A.; Finetti, R.; Niccolai, P.; Trezza, A.; Spiga, O.; Santucci, A.; Niccolai, N. Profiling of Protein-Coding Missense Mutations in Mendelian Rare Diseases: Clues from Structural Bioinformatics. Int. J. Mol. Sci. 2025, 26, 4072. https://doi.org/10.3390/ijms26094072
Visibelli A, Finetti R, Niccolai P, Trezza A, Spiga O, Santucci A, Niccolai N. Profiling of Protein-Coding Missense Mutations in Mendelian Rare Diseases: Clues from Structural Bioinformatics. International Journal of Molecular Sciences. 2025; 26(9):4072. https://doi.org/10.3390/ijms26094072
Chicago/Turabian StyleVisibelli, Anna, Rebecca Finetti, Piero Niccolai, Alfonso Trezza, Ottavia Spiga, Annalisa Santucci, and Neri Niccolai. 2025. "Profiling of Protein-Coding Missense Mutations in Mendelian Rare Diseases: Clues from Structural Bioinformatics" International Journal of Molecular Sciences 26, no. 9: 4072. https://doi.org/10.3390/ijms26094072
APA StyleVisibelli, A., Finetti, R., Niccolai, P., Trezza, A., Spiga, O., Santucci, A., & Niccolai, N. (2025). Profiling of Protein-Coding Missense Mutations in Mendelian Rare Diseases: Clues from Structural Bioinformatics. International Journal of Molecular Sciences, 26(9), 4072. https://doi.org/10.3390/ijms26094072