
Identification and Assessment of lncRNAs and mRNAs in PM2.5-Induced Hepatic Steatosis
 ,
,  and
and 
Abstract
1. Introduction
2. Results
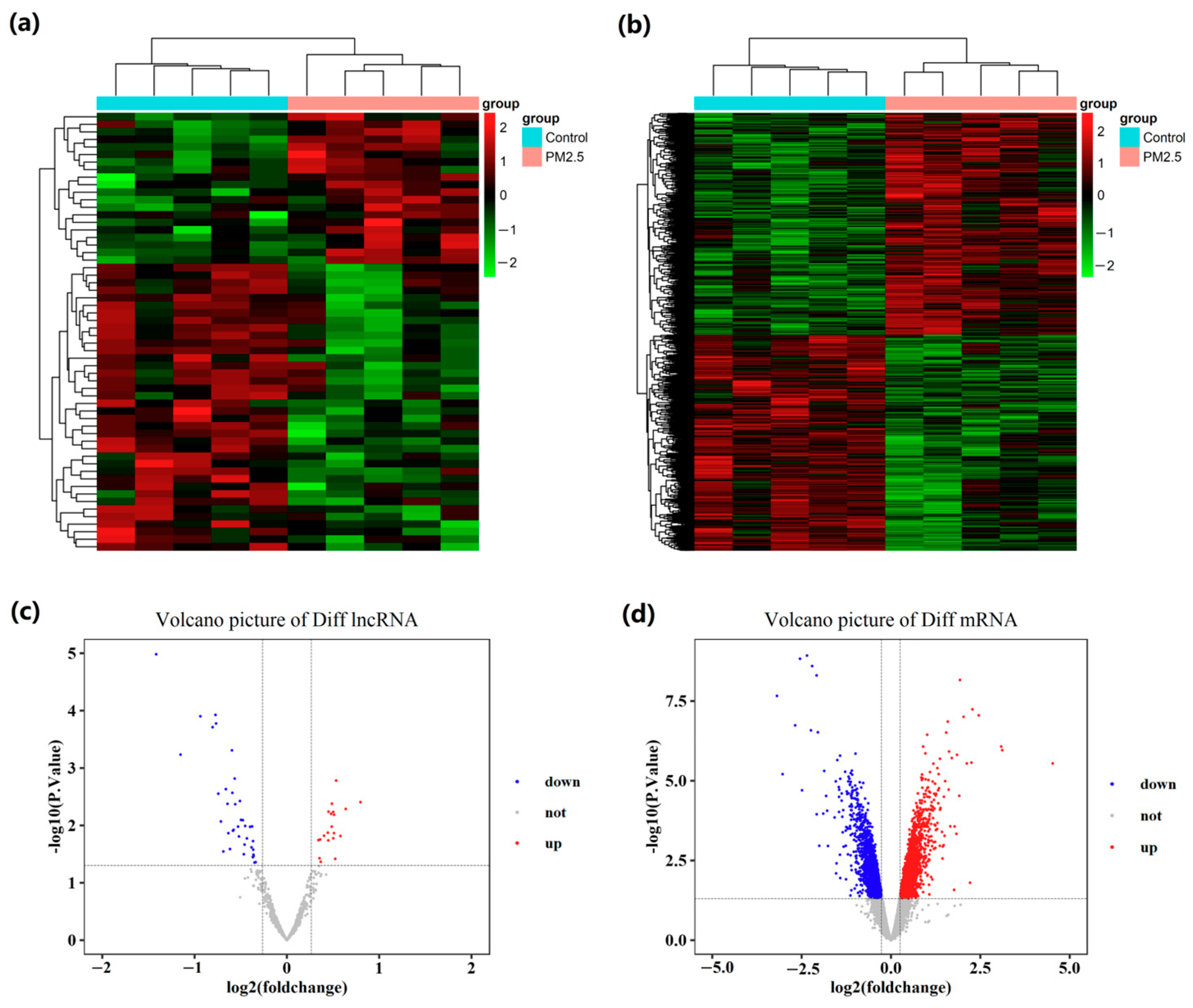
2.1. Global Differentially Expressed Gene Patterns in Liver Tissue
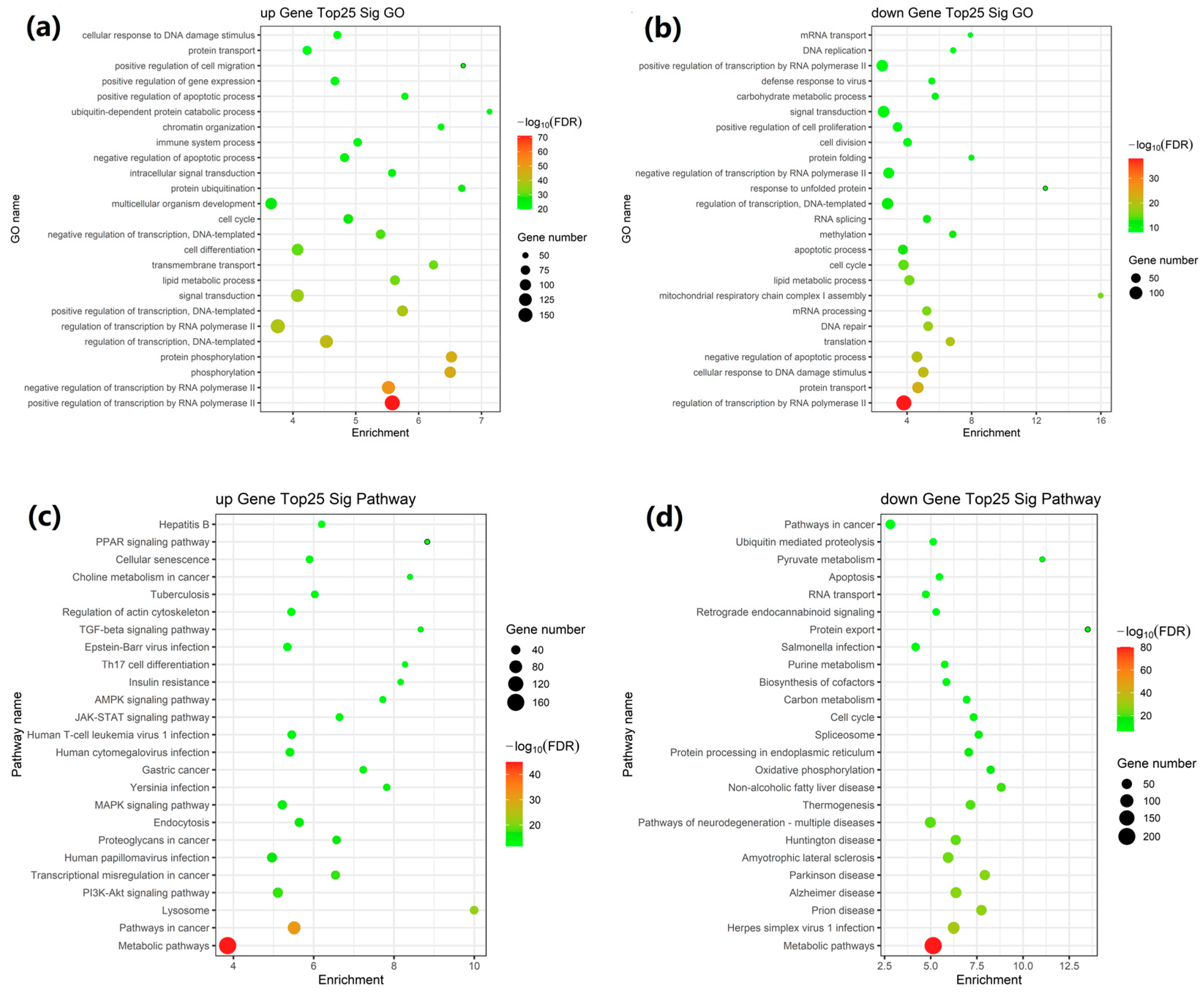
2.2. GO and KEGG Pathway Enrichment Analysis of DEGs
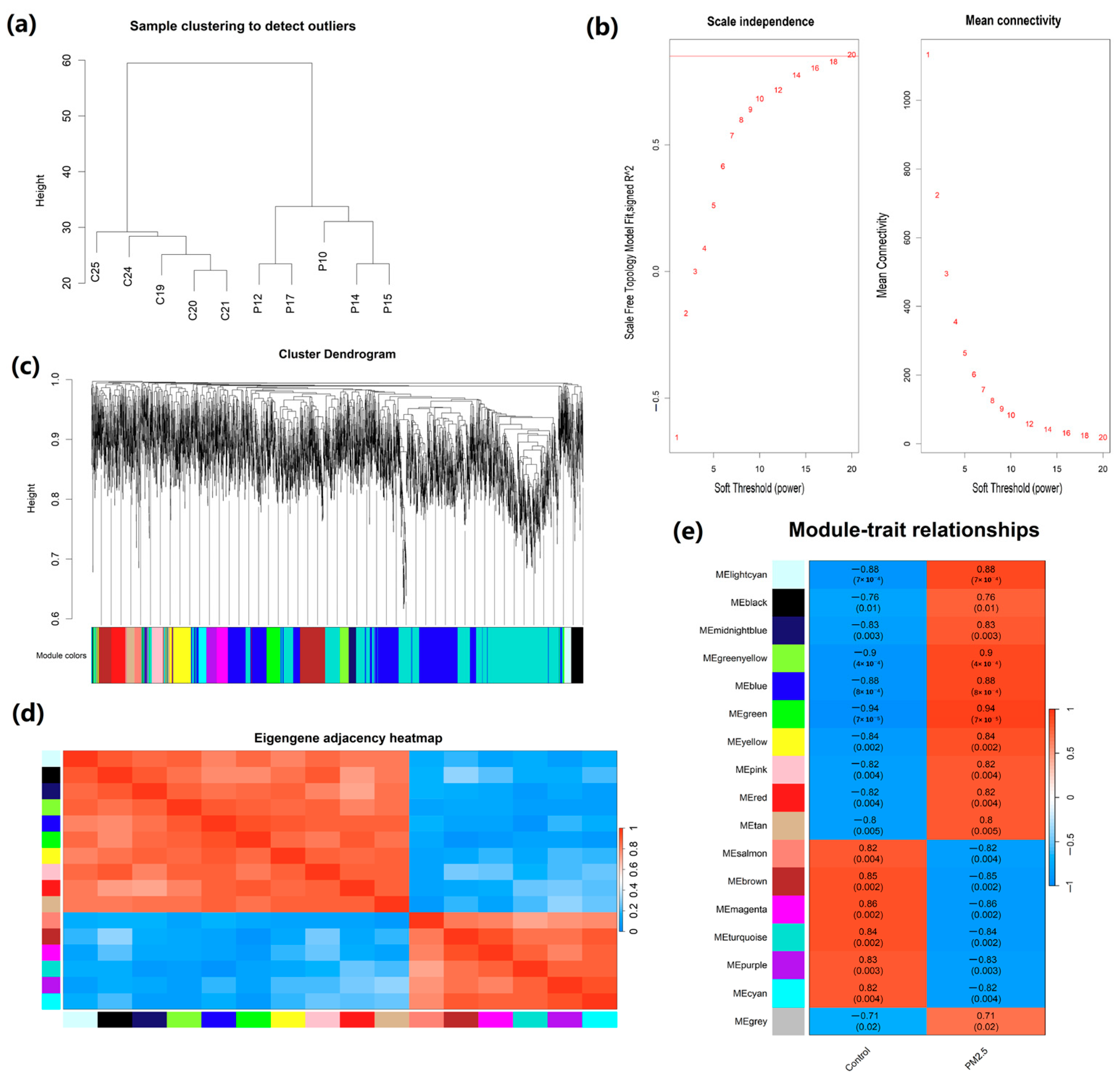
2.3. Construction of Weighted Co-Expression Network and Identification of Crucial Modules
2.4. Correlation Analysis Between Modules and Traits
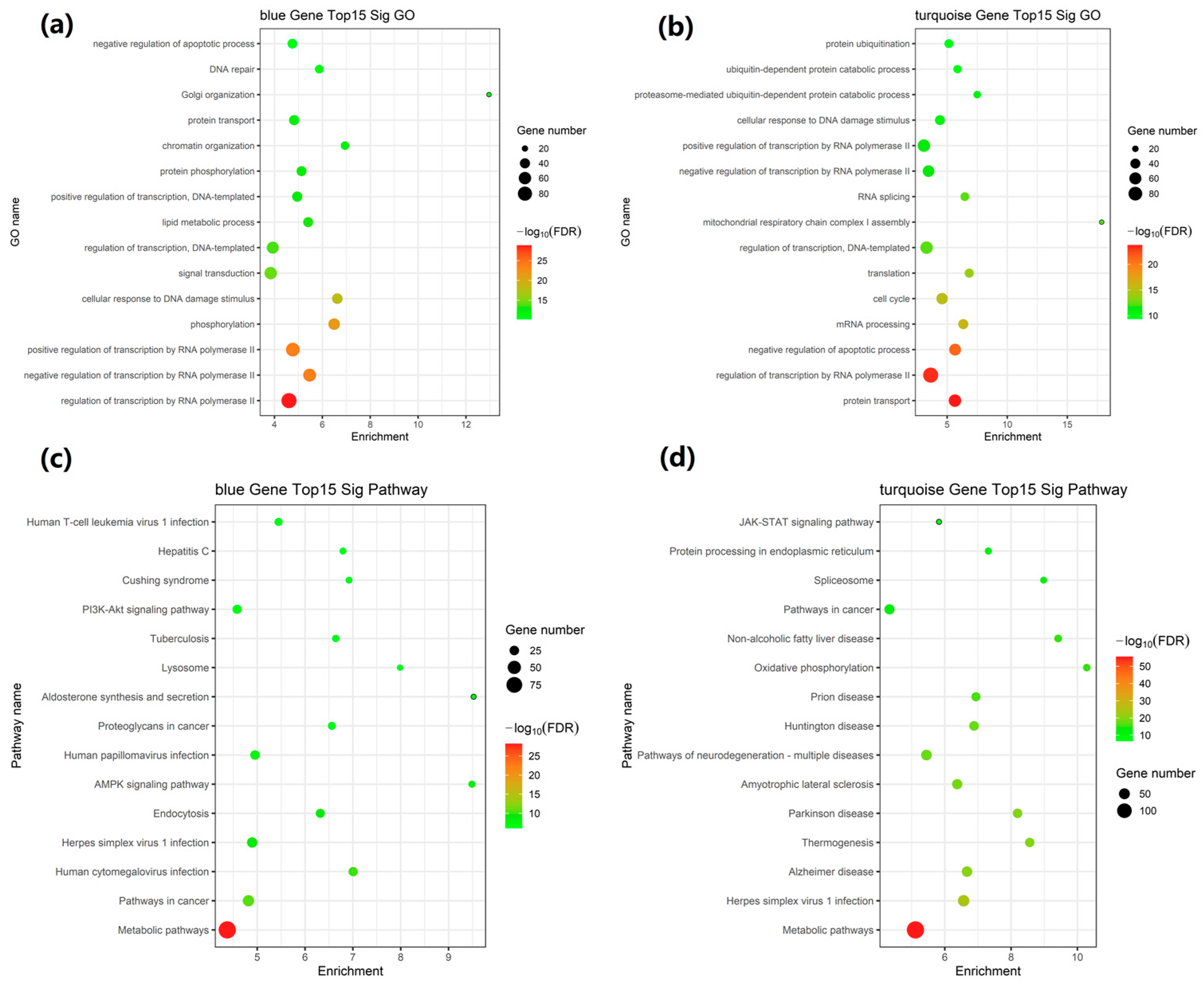
2.5. GO and KEGG Pathway Enrichment Analysis of Network Modules
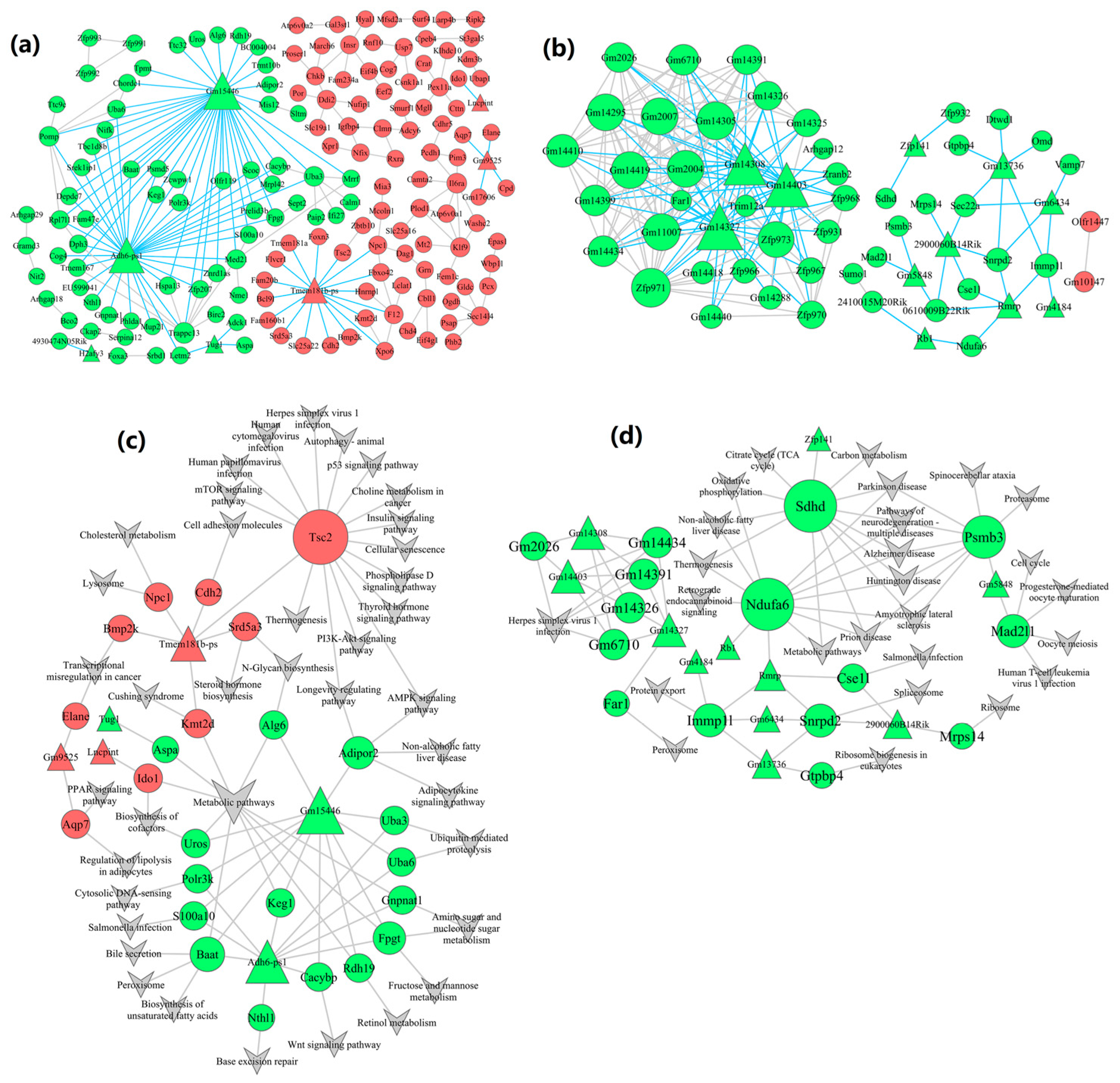
2.6. Construction of lncRNA-mRNA-Nets
2.7. Construction of lncRNA-mRNA-Pathway-Nets
2.8. Construction of Protein–Protein Interaction Network
3. Discussion
4. Materials and Methods
4.1. Data Retrieving and Processing
4.2. Analysis of Differentially Expressed Genes
4.3. GO and Pathway Enrichment Analysis of DEGs
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.5. Functional Enrichment Analysis
4.6. Construction of lncRNA-mRNA-Nets and lncRNA-mRNA-Pathway-Nets
4.7. Construction of Protein–Protein Interaction (PPI) Network
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PM2.5 | Fine particulate matter |
| WGCNA | Weighted gene co-expression network analysis |
| NAFLD | Non-alcoholic fatty liver disease |
| lncRNAs | Long noncoding RNAs |
| mRNAs | messenger RNAs |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DEGs | Differentially expressed genes |
| HCC | Hepatocellular carcinoma |
| β | Soft threshold power |
| FDR | False discovery rate |
| FC | Fold change |
| PPI | Protein–protein interaction |
| OR | Odds ratio |
| HR | Hazard ratios |
| AED | Aerodynamic diameter |
References
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharm. Ther. 2011, 34, 274–285. [Google Scholar]
- Fan, J.G. Epidemiology of alcoholic and nonalcoholic fatty liver disease in China. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S1), 11–17. [Google Scholar]
- Apte, J.S.; Marshall, J.D.; Cohen, A.J.; Brauer, M. Addressing Global Mortality from Ambient PM2.5. Environ. Sci. Technol. 2015, 49, 8057–8066. [Google Scholar] [PubMed]
- Mukherjee, A.; Agrawal, M. A Global Perspective of Fine Particulate Matter Pollution and Its Health Effects. Rev. Environ. Contam. Toxicol. 2018, 244, 5–51. [Google Scholar]
- Pai, S.J.; Carter, T.S.; Heald, C.L.; Kroll, J.H. Updated World Health Organization Air Quality Guidelines Highlight the Importance of Non-anthropogenic PM 2.5. Environ. Sci. Technol. Lett. 2022, 9, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, L.; Yang, G.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Wang, N. The influence of PM 2.5 exposure on non-alcoholic fatty liver disease. Life Sci. 2021, 270, 119135. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, S.; Bai, Q.; Pan, M.; Jiang, Y.; Liu, W.; Li, W.; Gong, Y.; Li, X. Air pollution exposure and prevalence of non-alcoholic fatty liver disease and related cirrhosis: A systematic review and meta-analysis. Ecotoxicol. Environ. Safe 2025, 289, 117469. [Google Scholar]
- Deng, P.; Tang, H.; Zhu, L.; Duan, J.; Li, F.; Li, Y.; Wang, J.; Wu, J.; Meng, C.; Wang, W.; et al. Association of long-term ambient fine particulate matter (PM2.5) and incident non-alcoholic fatty liver disease in Chinese adults. Environ. Pollut. 2023, 329, 121666. [Google Scholar]
- Aimuzi, R.; Xie, Z.; Qu, Y.; Luo, K.; Jiang, Y. Proteomic signatures of ambient air pollution and risk of non-alcoholic fatty liver disease: A prospective cohort study in the UK Biobank. Sci. Total. Environ. 2024, 957, 177529. [Google Scholar]
- Xu, M.X.; Ge, C.X.; Qin, Y.T.; Gu, T.T.; Lou, D.S.; Li, Q.; Hu, L.F.; Feng, J.; Huang, P.; Tan, J. Prolonged PM2.5 exposure elevates risk of oxidative stress-driven nonalcoholic fatty liver disease by triggering increase of dyslipidemia. Free Radic. Biol. Med. 2019, 130, 542–556. [Google Scholar]
- Lin, L.; Tian, L.; Li, T.; Sun, M.; Duan, J.; Yu, Y.; Sun, Z. Microarray analysis of mRNA expression profiles in liver of ob/ob mice with real-time atmospheric PM(2.5) exposure. Environ. Sci. Pollut. Res. 2022, 29, 76816–76832. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Pan, D.; Yu, J.; Wang, Y.; Sun, G.; Xia, H. Identification and Evaluation of Hub Long Noncoding RNAs and mRNAs in High Fat Diet Induced Liver Steatosis. Nutrients 2023, 15, 948. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.; Gao, Y.; Wang, Z.; Jin, F. Tussilagone protects acute lung injury from PM2.5 via alleviating Hif-1alpha/NF-kappaB-mediated inflammatory response. Environ. Toxicol. 2022, 37, 1198–1210. [Google Scholar]
- Ning, J.; Du, H.; Zhang, Y.; Liu, Q.; Jiang, T.; Pang, Y.; Tian, X.; Yan, L.; Niu, Y.; Zhang, R. N6-Methyladenosine Modification of CDH1 mRNA Promotes PM2.5-Induced Pulmonary Fibrosis via Mediating Epithelial Mesenchymal Transition. Toxicol. Sci. 2022, 185, 143–157. [Google Scholar]
- Brandt, A.; Kopp, F. Long Noncoding RNAs in Diet-Induced Metabolic Diseases. Int. J. Mol. Sci. 2024, 25, 5678. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chang, Z.; Wu, J.; Li, W. Air pollution and lung cancer incidence in China: Who are faced with a greater effect ? Environ. Int. 2019, 132, 105077. [Google Scholar]
- Zeng, Q.; Liu, C.H.; Wu, D.; Jiang, W.; Zhang, N.; Tang, H. LncRNA and circRNA in Patients with Non-Alcoholic Fatty Liver Disease: A Systematic Review. Biomolecules 2023, 13, 560. [Google Scholar] [CrossRef]
- Herman, A.B.; Tsitsipatis, D.; Gorospe, M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol. Cell 2022, 82, 2252–2266. [Google Scholar] [PubMed]
- Abdi, E.; Latifi-Navid, S.; Panahi, A.; Latifi-Navid, H. LncRNA polymorphisms and lung cancer risk. Pers. Med. 2023, 20, 511–522. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Qi, G.; Kong, W.; Mou, X.; Wang, S. A new method for excavating feature lncRNA in lung adenocarcinoma based on pathway crosstalk analysis. J. Cell. Biochem. 2019, 120, 9034–9046. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ho, S.; Sun, W.; Feng, P.; Lin, C.; Chen, K.; Chuang, H.; Tseng, C.; Chen, T.; Wu, S. Lnc-IL7R alleviates PM 2.5 -mediated cellular senescence and apoptosis through EZH2 recruitment in chronic obstructive pulmonary disease. Cell Biol. Toxicol. 2022, 38, 1097–1120. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, L.; Zhu, M.; Cheng, H. Effects and early diagnostic value of lncRNA H19 on sepsis-induced acute lung injury. Exp. Ther. Med. 2022, 23, 279. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, S.; Cui, Z.; Li, S.; Chen, Y.; Yin, Y.; Yin, Z. The relationship between LncRNAs and lung adenocarcinoma as well as their ceRNA network. Cancer Biomark. 2021, 31, 165–176. [Google Scholar] [CrossRef]
- Li, F.R.; Liao, J.; Zhu, B.; Li, X.; Cheng, Z.; Jin, C.; Mo, C.; Wu, X.; Li, Q.; Liang, F. Long-term exposure to air pollution and incident non-alcoholic fatty liver disease and cirrhosis: A cohort study. Liver Int. 2023, 43, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Wade, H.; Pan, K.; Zhang, B.; Zheng, W.; Su, Q. Mechanistic role of long non-coding RNAs in the pathogenesis of metabolic dysfunction-associated steatotic liver disease and fibrosis. eGastroenterology 2024, 2, e100115. [Google Scholar] [CrossRef]
- Ding, D.; Ye, G.; Lin, Y.; Lu, Y.; Zhang, H.; Zhang, X.; Hong, Z.; Huang, Q.; Chi, Y.; Chen, J.; et al. MicroRNA-26a-CD36 signaling pathway: Pivotal role in lipid accumulation in hepatocytes induced by PM 2.5 liposoluble extracts. Environ. Pollut. 2019, 248, 269–278. [Google Scholar] [CrossRef]
- Liu, C.; Xu, X.; Bai, Y.; Wang, T.; Rao, X.; Wang, A.; Sun, L.; Ying, Z.; Gushchina, L.; Maiseyeu, A.; et al. Air Pollution-Mediated Susceptibility to Inflammation and Insulin Resistance: Influence of CCR2 Pathways in Mice. Environ. Health Perspect. 2014, 122, 17–26. [Google Scholar] [CrossRef]
- Deng, X.; Feng, N.; Zheng, M.; Ye, X.; Lin, H.; Yu, X.; Gan, Z.; Fang, Z.; Zhang, H.; Gao, M.; et al. PM 2.5 exposure-induced autophagy is mediated by lncRNA loc146880 which also promotes the migration and invasion of lung cancer cells. Biochim. Biophys. Acta BBA-Gen. Subj. 2017, 1861, 112–125. [Google Scholar] [CrossRef]
- Spitz, A.Z.; Gavathiotis, E. Physiological and pharmacological modulation of BAX. Trends Pharmacol. Sci. 2022, 43, 206–220. [Google Scholar] [PubMed]
- Nangraj, A.S.; Selvaraj, G.; Kaliamurthi, S.; Kaushik, A.C.; Cho, W.C.; Wei, D.Q. Integrated PPI- and WGCNA-Retrieval of Hub Gene Signatures Shared Between Barrett’s Esophagus and Esophageal Adenocarcinoma. Front. Pharmacol. 2020, 11, 881. [Google Scholar]
- Xu, M.; Zhou, H.; Hu, P.; Pan, Y.; Wang, S.; Liu, L.; Liu, X. Identification and validation of immune and oxidative stress-related diagnostic markers for diabetic nephropathy by WGCNA and machine learning. Front. Immunol. 2023, 14, 1084531. [Google Scholar]
- Li, Y.; Yu, J.; Li, R.; Zhou, H.; Chang, X. New insights into the role of mitochondrial metabolic dysregulation and immune infiltration in septic cardiomyopathy by integrated bioinformatics analysis and experimental validation. Cell. Mol. Biol. Lett. 2024, 29, 21. [Google Scholar] [CrossRef]
- Sui, J.; Zhang, Y.; Zhang, L.; Xia, H. Identification and Evaluation of Hub Long Non-Coding RNAs and mRNAs in PM2.5-Induced Lung Cell Injury. Int. J. Mol. Sci. 2025, 26, 911. [Google Scholar] [CrossRef]
- Zhou, B.; Wu, Y.; Cheng, P.; Wu, C. Long noncoding RNAs with peptide-encoding potential identified in esophageal squamous cell carcinoma: KDM4A—AS1—Encoded peptide weakens cancer cell viability and migratory capacity. Mol. Oncol. 2023, 17, 1419–1436. [Google Scholar]
- Chen, T.; Meng, Y.; Zhou, Z.; Li, H.; Wan, L.; Kang, A.; Guo, W.; Ren, K.; Song, X.; Chen, Y.; et al. GAS5 protects against nonalcoholic fatty liver disease via miR-28a-5p/MARCH7/NLRP3 axis-mediated pyroptosis. Cell Death Differ. 2023, 30, 1829–1848. [Google Scholar]
- Sun, H.; Kemper, J.K. MicroRNA regulation of AMPK in nonalcoholic fatty liver disease. Exp. Mol. Med. 2023, 55, 1974–1981. [Google Scholar]
- Zhang, Z.; Wang, S.; Zhu, Z.; Nie, B. Identification of potential feature genes in non-alcoholic fatty liver disease using bioinformatics analysis and machine learning strategies. Comput. Biol. Med. 2023, 157, 106724. [Google Scholar]
- Guo, J.; Fang, W.; Sun, L.; Lu, Y.; Dou, L.; Huang, X.; Tang, W.; Yu, L.; Li, J. Ultraconserved element uc.372 drives hepatic lipid accumulation by suppressing miR-195/miR4668 maturation. Nat. Commun. 2018, 9, 612. [Google Scholar]
- Zhang, Y.; Sun, J.; Yao, H.; Lin, Y.; Wei, J.; Hu, G.; Guo, J.; Li, J. Ultraconserved element uc.333 increases insulin sensitivity by binding to miR-223. Aging 2020, 12, 6667–6679. [Google Scholar] [CrossRef]
- Zhang, H.; Niu, Q.; Liang, K.; Li, X.; Jiang, J.; Bian, C. Effect of LncPVT1/miR-20a-5p on Lipid Metabolism and Insulin Resistance in NAFLD. Diabetes Metab. Syndr. Obes. 2021, 14, 4599–4608. [Google Scholar] [PubMed]
- Albadawy, R.; Agwa, S.; Khairy, E.; Saad, M.; El, T.N.; Othman, M.; Matboli, M. Clinical Significance of HSPD1/MMP14/ITGB1/miR-6881-5P/Lnc-SPARCL1-1:2 RNA Panel in NAFLD/NASH Diagnosis: Egyptian Pilot Study. Biomedicinaes 2021, 9, 1248. [Google Scholar]
- Hu, M.J.; Long, M.; Dai, R.J. Acetylation of H3K27 activated lncRNA NEAT1 and promoted hepatic lipid accumulation in non-alcoholic fatty liver disease via regulating miR-212-5p/GRIA3. Mol. Cell. Biochem. 2022, 477, 191–203. [Google Scholar] [PubMed]
- Zhou, W.; Qiu, K. The correlation between lncRNA NEAT1 and serum hepcidin in the peripheral blood of non-alcoholic fatty liver disease patients. Am. J. Transl. Res. 2022, 14, 2593–2599. [Google Scholar]
- Meng, X.; Long, M.; Yue, N.; Li, Q.; Chen, J.; Zhao, H.; Deng, W. LncRNA MEG3 Restrains Hepatic Lipogenesis via the FOXO1 Signaling Pathway in HepG2 Cells. Cell Biochem. Biophys. 2024, 82, 1253–1259. [Google Scholar] [PubMed]
- Yin, J.; Chen, X.; Zhang, F.; Zhao, M. RMRP inhibition prevents NAFLD progression in rats via regulating miR-206/PTPN1 axis. Mamm. Genome 2022, 33, 480–489. [Google Scholar] [PubMed]
- Zhou, N.; He, Z.; Tang, H.; Jiang, B.; Cheng, W. LncRNA RMRP/miR-613 axis is associated with poor prognosis and enhances the tumorigenesis of hepatocellular carcinoma by impacting oncogenic phenotypes. Am. J. Transl. Res. 2019, 11, 2801–2815. [Google Scholar]
- Tang, L.; Wang, Y.; Wang, H.; Xu, B.; Ji, H.; Xu, G.; Ge, X.; Li, Q.; Miao, L. Long noncoding-RNA component of mitochondrial RNA processing endoribonuclease is involved in the progression of cholangiocarcinoma by regulating microRNA-217. Cancer Sci. 2019, 110, 2166–2179. [Google Scholar]
- Hongfeng, Z.; Andong, J.; Liwen, S.; Mingping, B.; Xiaowei, Y.; Mingyong, L.; Aimin, Y. lncRNA RMRP knockdown suppress hepatocellular carcinoma biological activities via regulation miRNA-206/TACR1. J. Cell. Biochem. 2020, 121, 1690–1702. [Google Scholar] [CrossRef]
- Shao, C.; Liu, G.; Zhang, X.; Li, A.; Guo, X. Long Noncoding RNA RMRP Suppresses the Tumorigenesis of Hepatocellular Carcinoma Through Targeting microRNA-766. OncoTargets Ther. 2020, 13, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Deregulation of RB1 expression by loss of imprinting in human hepatocellular carcinoma. J. Pathol. 2014, 233, 392–401. [Google Scholar] [CrossRef]
- Chand, V.; Liao, X.; Guzman, G.; Benevolenskaya, E.; Raychaudhuri, P. Hepatocellular carcinoma evades RB1-induced senescence by activating the FOXM1-FOXO1 axis. Oncogene 2022, 41, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Nwadiugwu, M.C. Expression, Interaction, and Role of Pseudogene Adh6-ps1 in Cancer Phenotypes. Bioinform. Biol. Insights 2021, 15, 11779322211040591. [Google Scholar] [CrossRef]
- Nicholson, L.; Piras, I.S.; DeBoth, M.D.; Siniard, A.; Heras-Garvin, A.; Stefanova, N.; Huentelman, M.J. Transcriptomic insights into multiple system atrophy from a PLP-alpha-synuclein transgenic mouse model. Brain Res. 2024, 1834, 148912. [Google Scholar] [CrossRef]
- Wang, C.Y.; Stapleton, D.S.; Schueler, K.L.; Rabaglia, M.E.; Oler, A.T.; Keller, M.P.; Kendziorski, C.M.; Broman, K.W.; Yandell, B.S.; Schadt, E.E.; et al. Tsc2, a positional candidate gene underlying a quantitative trait locus for hepatic steatosis. J. Lipid Res. 2012, 53, 1493–1501. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Yamamoto, S.; Tatsuno, K.; Renard-Guillet, C.; Tsuji, S.; Hayashi, A.; Ueda, H.; Fukuda, S.; Fujita, T.; Katoh, H.; et al. Accumulation of Molecular Aberrations Distinctive to Hepatocellular Carcinoma Progression. Cancer Res. 2020, 80, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, M.; Peng, R.; Zhang, Y.; Qiao, Z.; Sun, N. ACACA reduces lipid accumulation through dual regulation of lipid metabolism and mitochondrial function via AMPK–PPARα–CPT1A axis. J. Transl. Med. 2024, 22, 1–14. [Google Scholar] [CrossRef]
- Gangaraj, K.P.; Rajesh, M.K. Dataset of dual RNA-sequencing of Phytophthora palmivora infecting coconut (Cocos nucifera L.). Data Brief 2020, 30, 105455. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef]
- Xiao, K.W.; Li, J.L.; Zeng, Z.H.; Liu, Z.B.; Hou, Z.Q.; Yan, X.; Cai, L. Monocytes affect bone mineral density in pre-and postmenopausal women through ribonucleoprotein complex biogenesis by integrative bioinformatics analysis. Sci. Rep. 2019, 9, 17290. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Liang, Y.; Shi, J.; Xiao, L.; Tang, L.; Guo, Y.; Chen, F.; Lin, G. Identification and Application of a Novel Immune-Related lncRNA Signature on the Prognosis and Immunotherapy for Lung Adenocarcinoma. Diagnostics 2022, 12, 2891. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Number of All | Number of mRNAs | Number of lncRNAs |
|---|---|---|---|
| Turquoise | 1399 | 1379 | 20 |
| Blue | 1004 | 995 | 9 |
| Brown | 355 | 346 | 9 |
| Yellow | 175 | 170 | 5 |
| Green | 147 | 146 | 1 |
| Red | 109 | 108 | 1 |
| Black | 95 | 92 | 3 |
| Pink | 95 | 94 | 1 |
| Magenta | 93 | 91 | 2 |
| Purple | 88 | 87 | 1 |
| Greenyellow | 79 | 77 | 2 |
| Tan | 73 | 73 | 0 |
| Cyan | 69 | 68 | 1 |
| Salmon | 69 | 67 | 2 |
| Midnight blue | 68 | 68 | 0 |
| Light cyan | 63 | 63 | 0 |
| Grey | 4 | 3 | 1 |
| Module | Go Id | Go Name | Enrichment | p-Value |
|---|---|---|---|---|
| blue module | GO:0006357 | regulation of transcription by RNA polymerase II | 4.604618417 | 4.0808 × 10−33 |
| GO:0000122 | negative regulation of transcription by RNA polymerase II | 5.469731618 | 1.36261 × 10−27 | |
| GO:0045944 | positive regulation of transcription by RNA polymerase II | 4.766936394 | 1.36552 × 10−27 | |
| GO:0016310 | phosphorylation | 6.49228687 | 4.00222 × 10−25 | |
| GO:0006974 | cellular response to DNA damage stimulus | 6.62509856 | 1.2414 × 10−21 | |
| GO:0007165 | signal transduction | 3.841899685 | 6.64258 × 10−18 | |
| GO:0006355 | regulation of transcription, DNA-templated | 3.92919919 | 3.5163 × 10−17 | |
| GO:0006629 | lipid metabolic process | 5.406814483 | 1.19225 × 10−16 | |
| GO:0045893 | positive regulation of transcription, DNA-templated | 4.949471573 | 4.01724 × 10−16 | |
| GO:0006468 | protein phosphorylation | 5.130762335 | 6.86727 × 10−16 | |
| GO:0006325 | chromatin organization | 6.945588197 | 2.18748 × 10−15 | |
| GO:0015031 | protein transport | 4.820964444 | 2.30318 × 10−15 | |
| GO:0007030 | Golgi organization | 12.96249113 | 8.20478 × 10−14 | |
| GO:0006281 | DNA repair | 5.872692459 | 1.71088 × 10−13 | |
| GO:0043066 | negative regulation of the apoptotic process | 4.746593849 | 2.40923 × 10−13 | |
| turquoise module | GO:0015031 | protein transport | 5.64659069 | 4.72814 × 10−28 |
| GO:0006357 | regulation of transcription by RNA polymerase II | 3.62721593 | 2.76769 × 10−27 | |
| GO:0043066 | negative regulation of the apoptotic process | 5.658760066 | 4.18637 × 10−25 | |
| GO:0006397 | mRNA processing | 6.342185195 | 1.2486 × 10−19 | |
| GO:0007049 | cell cycle | 4.580467085 | 7.40711 × 10−19 | |
| GO:0006412 | translation | 6.837668413 | 2.47991 × 10−17 | |
| GO:0006355 | regulation of transcription, DNA-templated | 3.287217115 | 9.34773 × 10−16 | |
| GO:0032981 | mitochondrial respiratory chain complex I assembly | 17.86166443 | 1.08356 × 10−15 | |
| GO:0008380 | RNA splicing | 6.472296819 | 1.18584 × 10−15 | |
| GO:0000122 | negative regulation of transcription by RNA polymerase II | 3.449634515 | 9.34622 × 10−15 | |
| GO:0045944 | positive regulation of transcription by RNA polymerase II | 3.065161702 | 1.4967 × 10−14 | |
| GO:0006974 | cellular response to DNA damage stimulus | 4.398665041 | 8.9227 × 10−14 | |
| GO:0043161 | proteasome-mediated ubiquitin-dependent protein catabolic process | 7.501899059 | 9.65769 × 10−14 | |
| GO:0006511 | ubiquitin-dependent protein catabolic process | 5.86085864 | 1.08627 × 10−12 | |
| GO:0016567 | protein ubiquitination | 5.144327333 | 1.63207 × 10−12 |
| Module | Pathway Id | Pathway Name | Enrichment | p-Value |
|---|---|---|---|---|
| blue module | 01100 | Metabolic pathways | 4.373215837 | 2.05556 × 10−31 |
| 05200 | Pathways in cancer | 4.81652525 | 1.57133 × 10−13 | |
| 05163 | Human cytomegalovirus infection | 7.005463957 | 5.87512 × 10−13 | |
| 05168 | Herpes simplex virus 1 infection | 4.894647306 | 6.88501 × 10−12 | |
| 04144 | Endocytosis | 6.318653765 | 1.60792 × 10−11 | |
| 04152 | AMPK signaling pathway | 9.488882397 | 6.80369 × 10−11 | |
| 05165 | Human papillomavirus infection | 4.954140257 | 8.83013 × 10−10 | |
| 05205 | Proteoglycans in cancer | 6.561215023 | 1.8569 × 10−9 | |
| 04925 | Aldosterone synthesis and secretion | 9.523767994 | 5.10304 × 10−9 | |
| 04142 | Lysosomes | 7.985872399 | 1.24271 × 10−8 | |
| 05152 | Tuberculosis | 6.642217678 | 1.4244 × 10−8 | |
| 04151 | PI3K-Akt signaling pathway | 4.579244778 | 2.09365 × 10−8 | |
| 04934 | Cushing syndrome | 6.918976748 | 2.50236 × 10−8 | |
| 05160 | Hepatitis C | 6.79317717 | 3.21535 × 10−8 | |
| 05166 | Human T-cell leukemia virus 1 infection | 5.445542833 | 3.60359 × 10−8 | |
| turquoise Module | 01100 | Metabolic pathways | 5.115202324 | 8.71148 × 10−59 |
| 05168 | Herpes simplex virus 1 infection | 6.568939087 | 1.82711 × 10−27 | |
| 05010 | Alzheimer disease | 6.670896013 | 6.91349 × 10−23 | |
| 04714 | Thermogenesis | 8.561950013 | 3.59115 × 10−22 | |
| 05012 | Parkinson disease | 8.194128949 | 4.42746 × 10−22 | |
| 05014 | Amyotrophic lateral sclerosis | 6.374411746 | 3.97572 × 10−21 | |
| 05022 | Pathways of neurodegeneration—multiple diseases | 5.446956194 | 3.79844 × 10−20 | |
| 05016 | Huntington disease | 6.882950985 | 6.23685 × 10−20 | |
| 05020 | Prion disease | 6.939723166 | 4.91892 × 10−18 | |
| 00190 | Oxidative phosphorylation | 10.28220814 | 1.25791 × 10−17 | |
| 04932 | Non-alcoholic fatty liver disease | 9.418775033 | 2.63225 × 10−17 | |
| 05200 | Pathways in cancer | 4.331782567 | 7.20715 × 10−15 | |
| 03040 | Spliceosome | 8.980818214 | 2.52467 × 10−14 | |
| 04141 | Protein processing in endoplasmic reticulum | 7.314715047 | 5.75875 × 10−13 | |
| 04630 | JAK-STAT signaling pathway | 5.826179003 | 1.07188 × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, P.; Xia, H.; Li, X.; Wang, Y.; Hu, B.; Yang, Y.; Sun, G.; Sui, J. Identification and Assessment of lncRNAs and mRNAs in PM2.5-Induced Hepatic Steatosis. Int. J. Mol. Sci. 2025, 26, 2808. https://doi.org/10.3390/ijms26062808
Tian P, Xia H, Li X, Wang Y, Hu B, Yang Y, Sun G, Sui J. Identification and Assessment of lncRNAs and mRNAs in PM2.5-Induced Hepatic Steatosis. International Journal of Molecular Sciences. 2025; 26(6):2808. https://doi.org/10.3390/ijms26062808
Chicago/Turabian StyleTian, Peixuan, Hui Xia, Xinbao Li, Ying Wang, Bihuan Hu, Yu Yang, Guiju Sun, and Jing Sui. 2025. "Identification and Assessment of lncRNAs and mRNAs in PM2.5-Induced Hepatic Steatosis" International Journal of Molecular Sciences 26, no. 6: 2808. https://doi.org/10.3390/ijms26062808
APA StyleTian, P., Xia, H., Li, X., Wang, Y., Hu, B., Yang, Y., Sun, G., & Sui, J. (2025). Identification and Assessment of lncRNAs and mRNAs in PM2.5-Induced Hepatic Steatosis. International Journal of Molecular Sciences, 26(6), 2808. https://doi.org/10.3390/ijms26062808