
Seco-Duocarmycin SA in Aggressive Glioblastoma Cell Lines
 , , and
, , and
Abstract
1. Introduction
2. Results
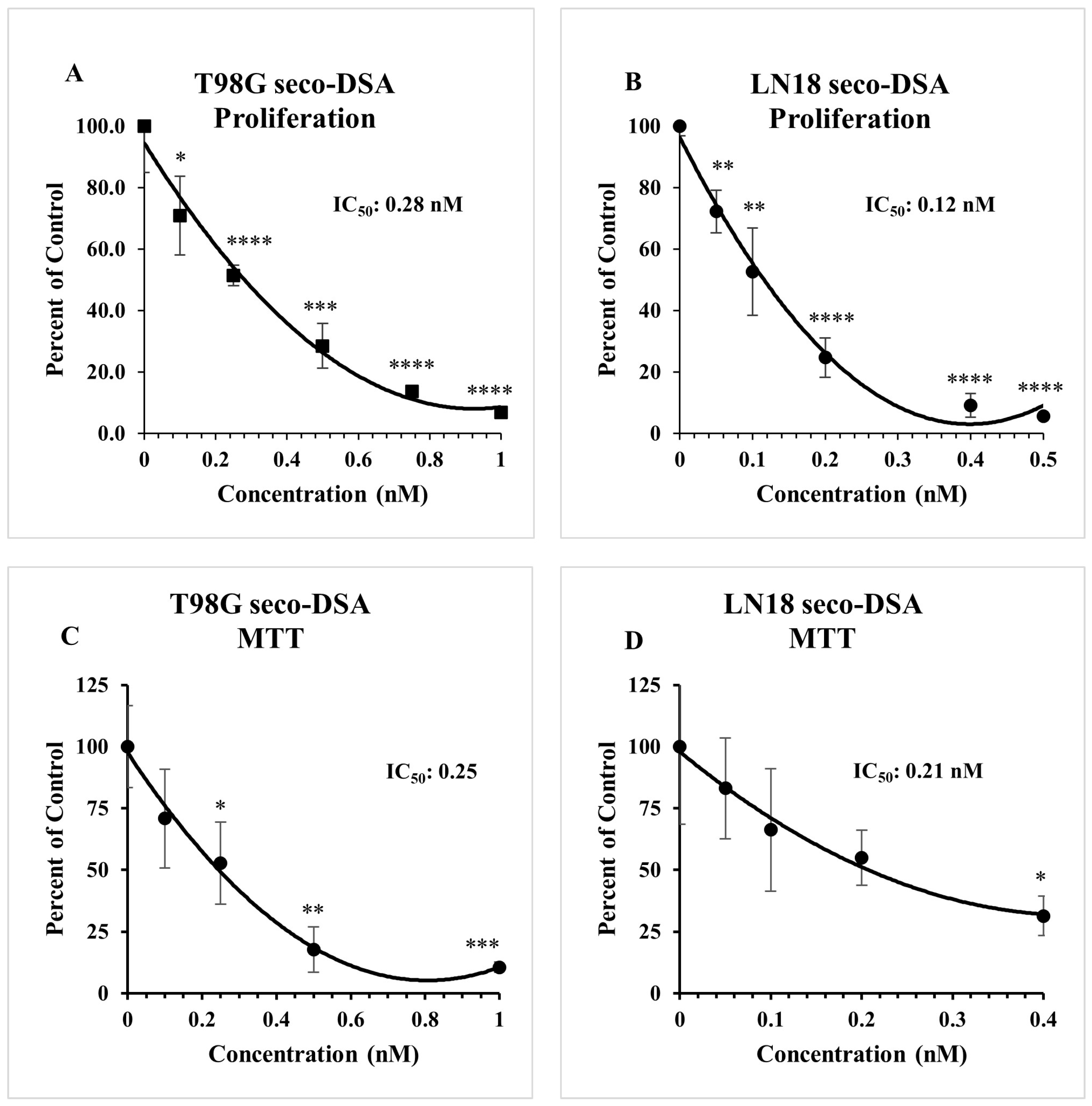
2.1. Both DSA and Seco-DSA Show Significant Potency in GBM Cell Lines
2.2. Seco-DSA Inhibits Cell Viability in GBM Cells
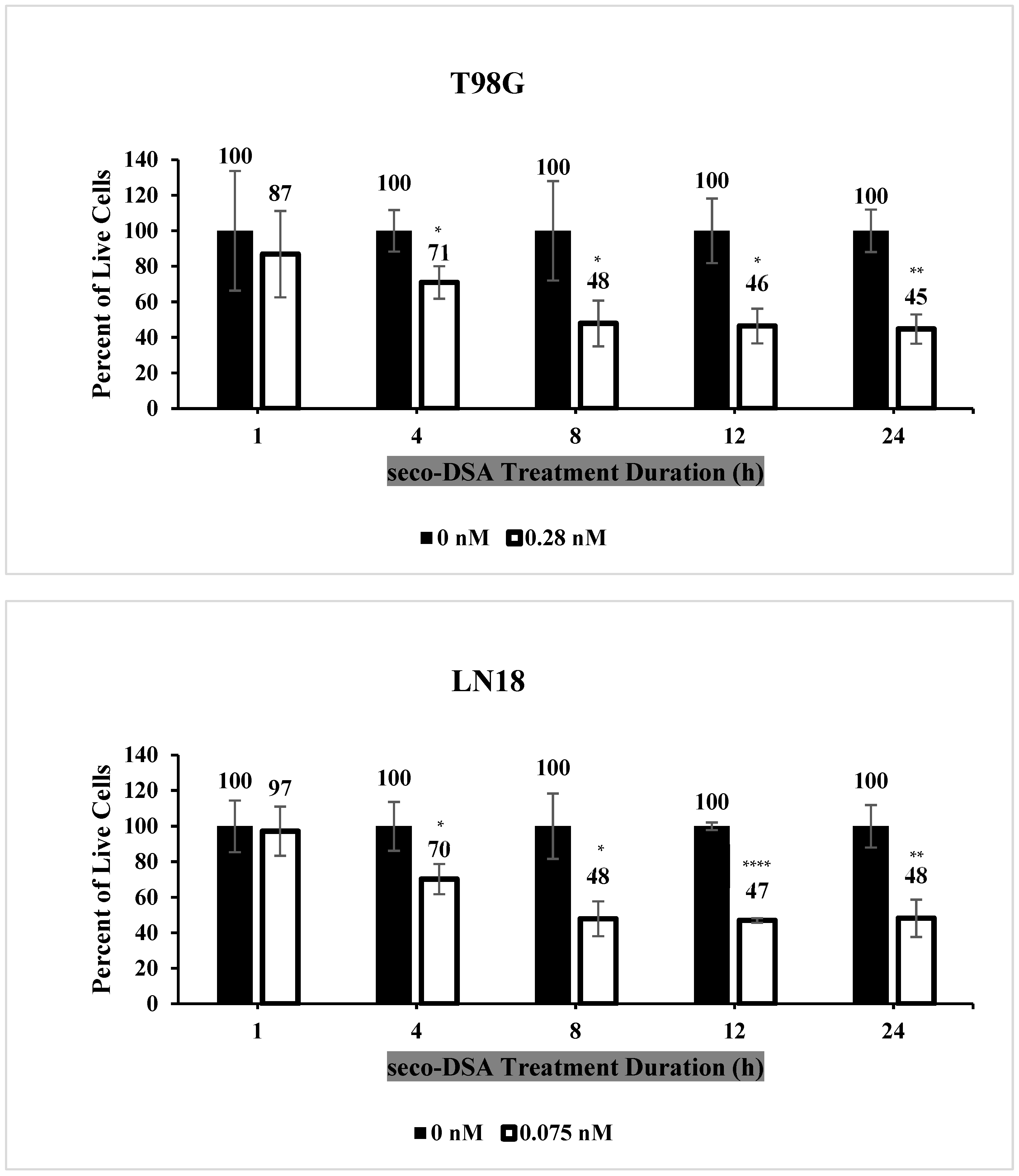
2.3. Seco-DSA Achieves Maximal Efficacy Within 8 h
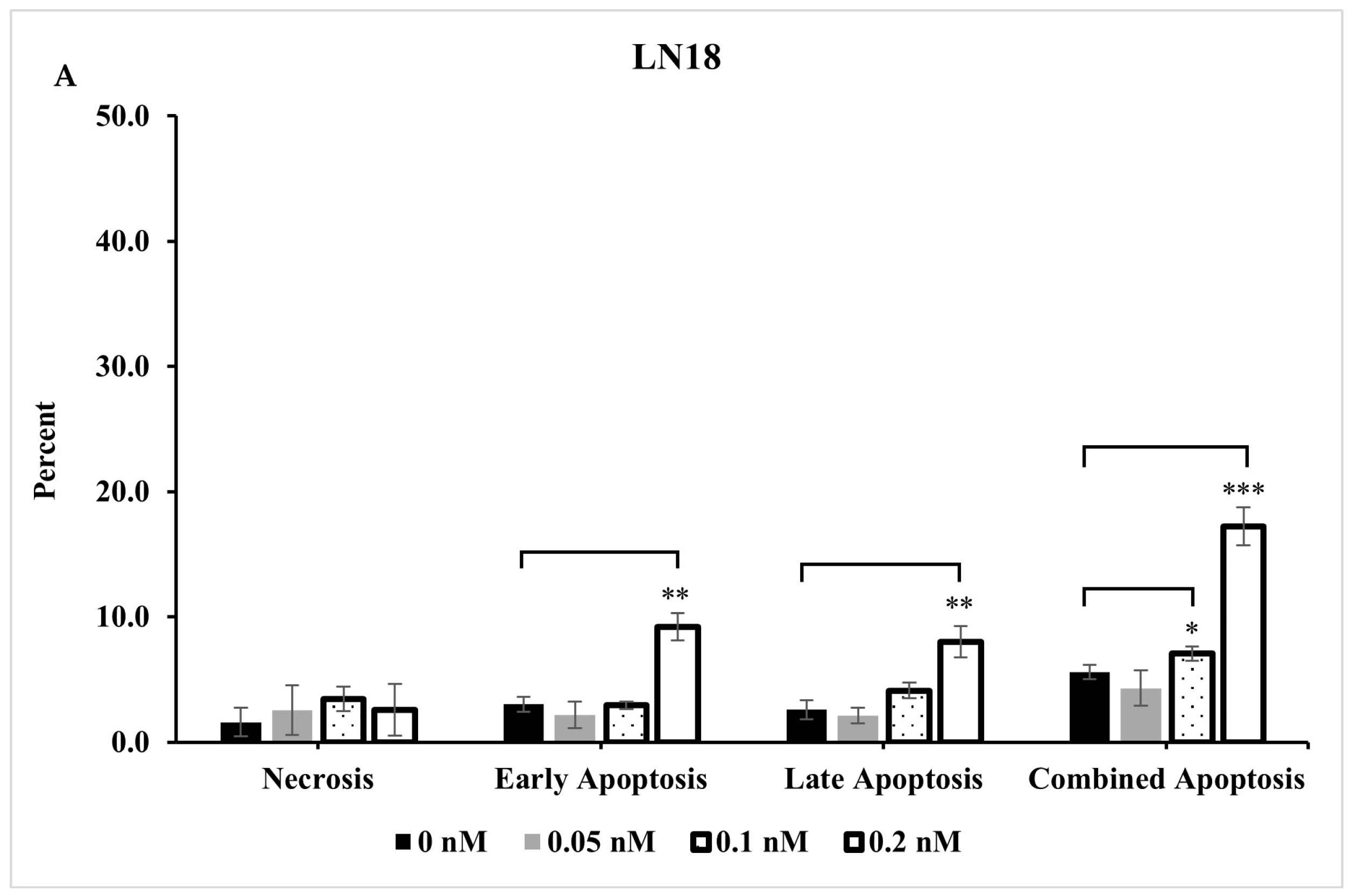
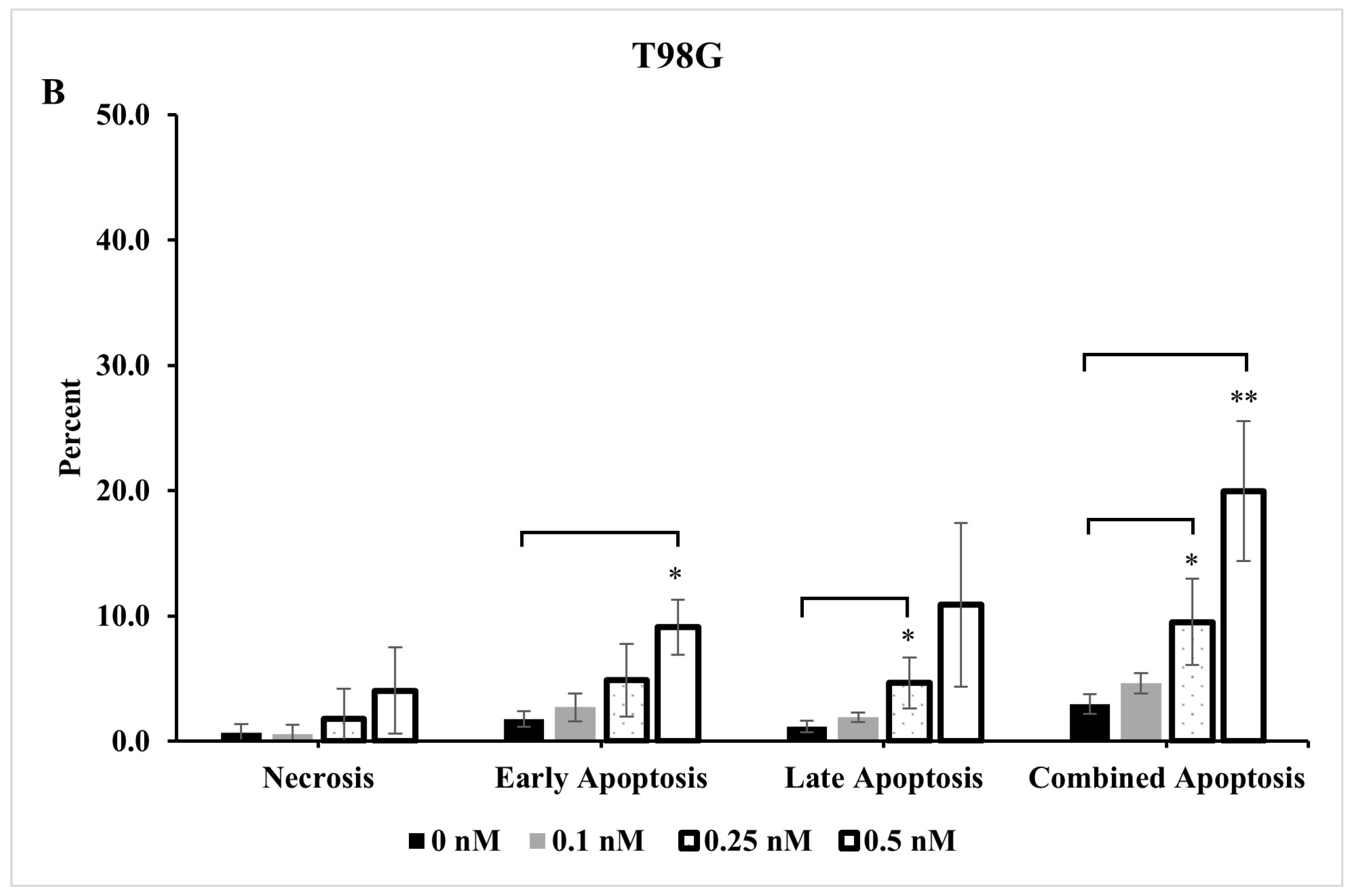
2.4. Extent of Apoptosis and Necrosis Does Not Fully Account for Significant Reduction Observed in Live Cells
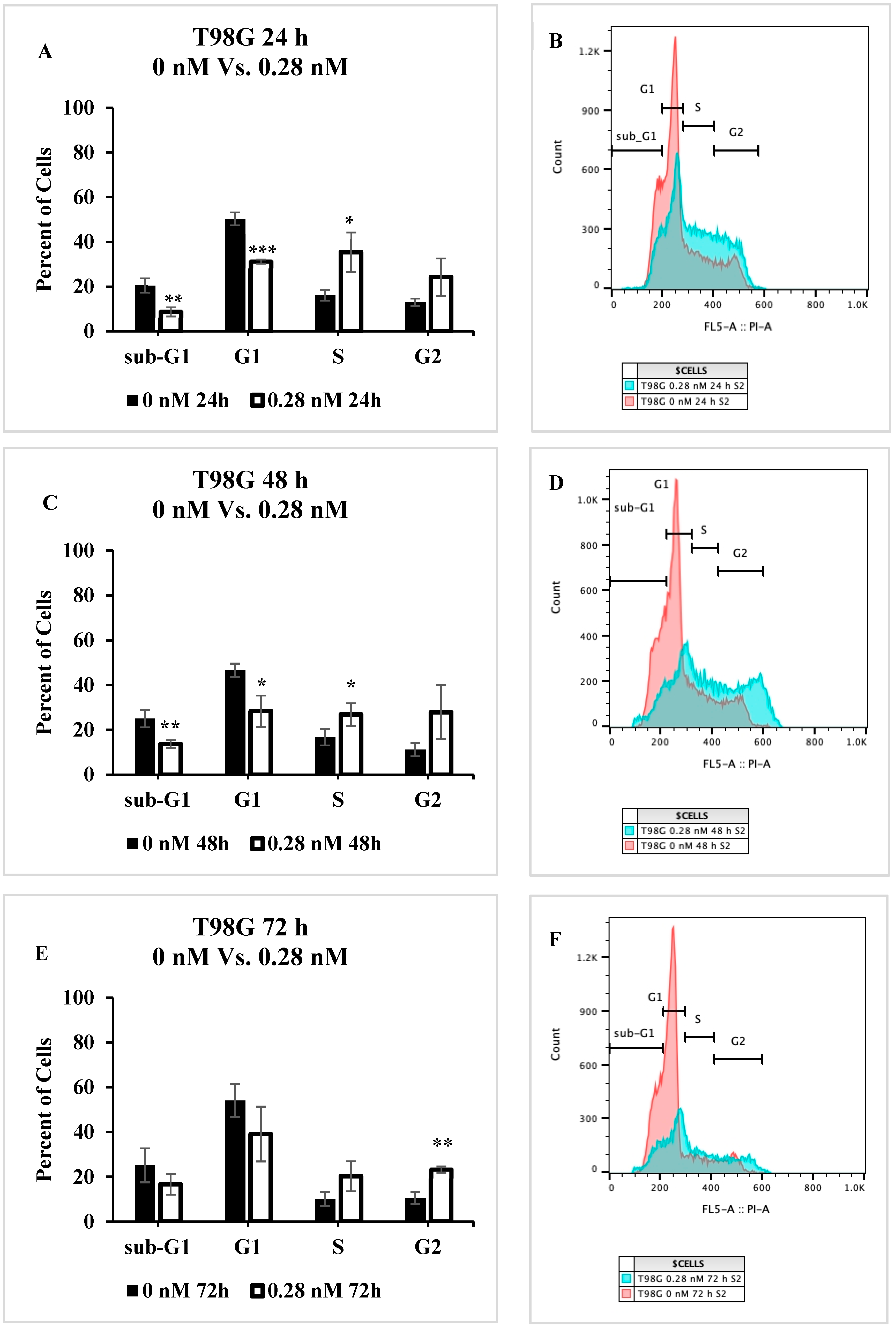
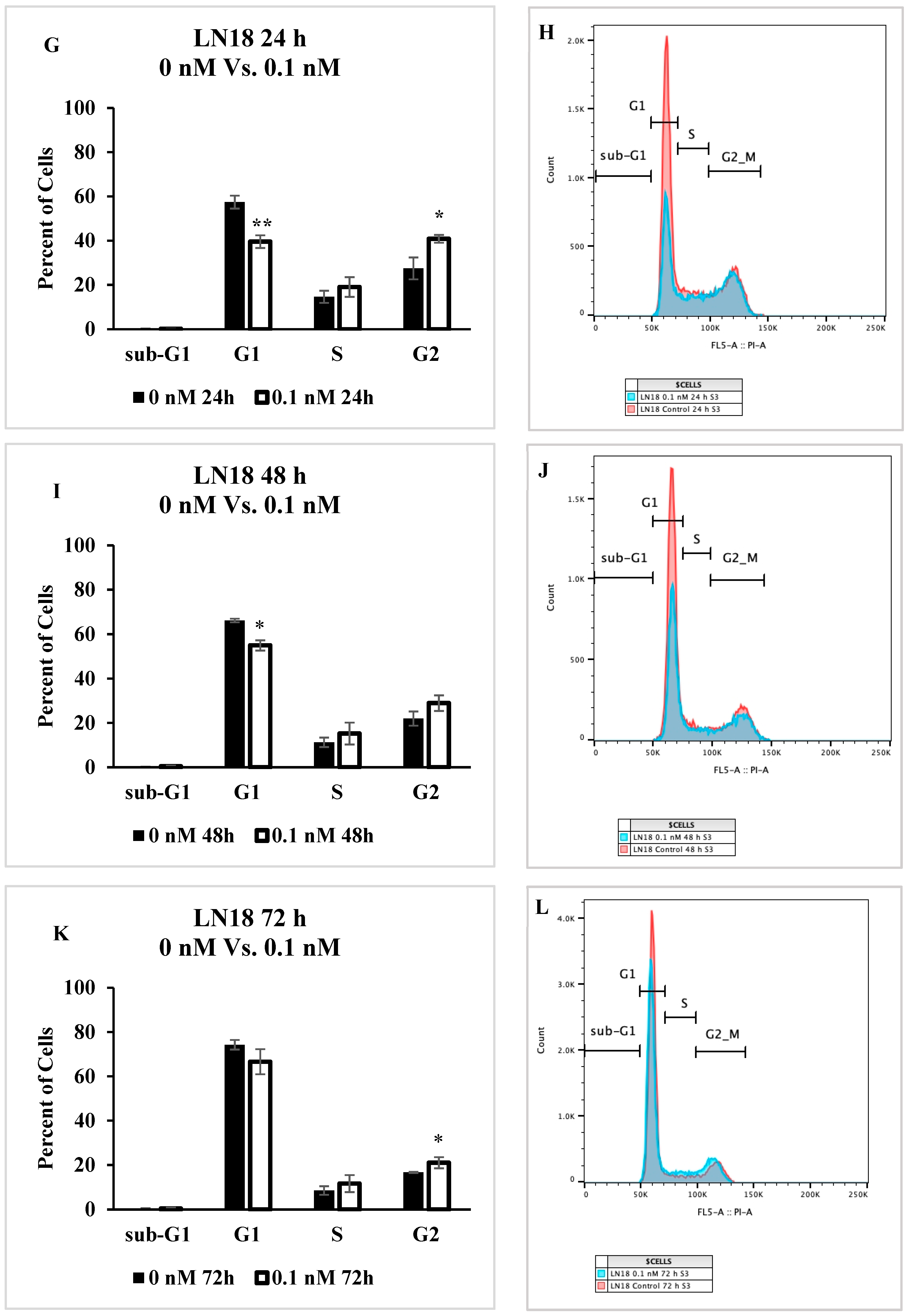
2.5. Cell Cycle Progression Is Impacted by Seco-DSA
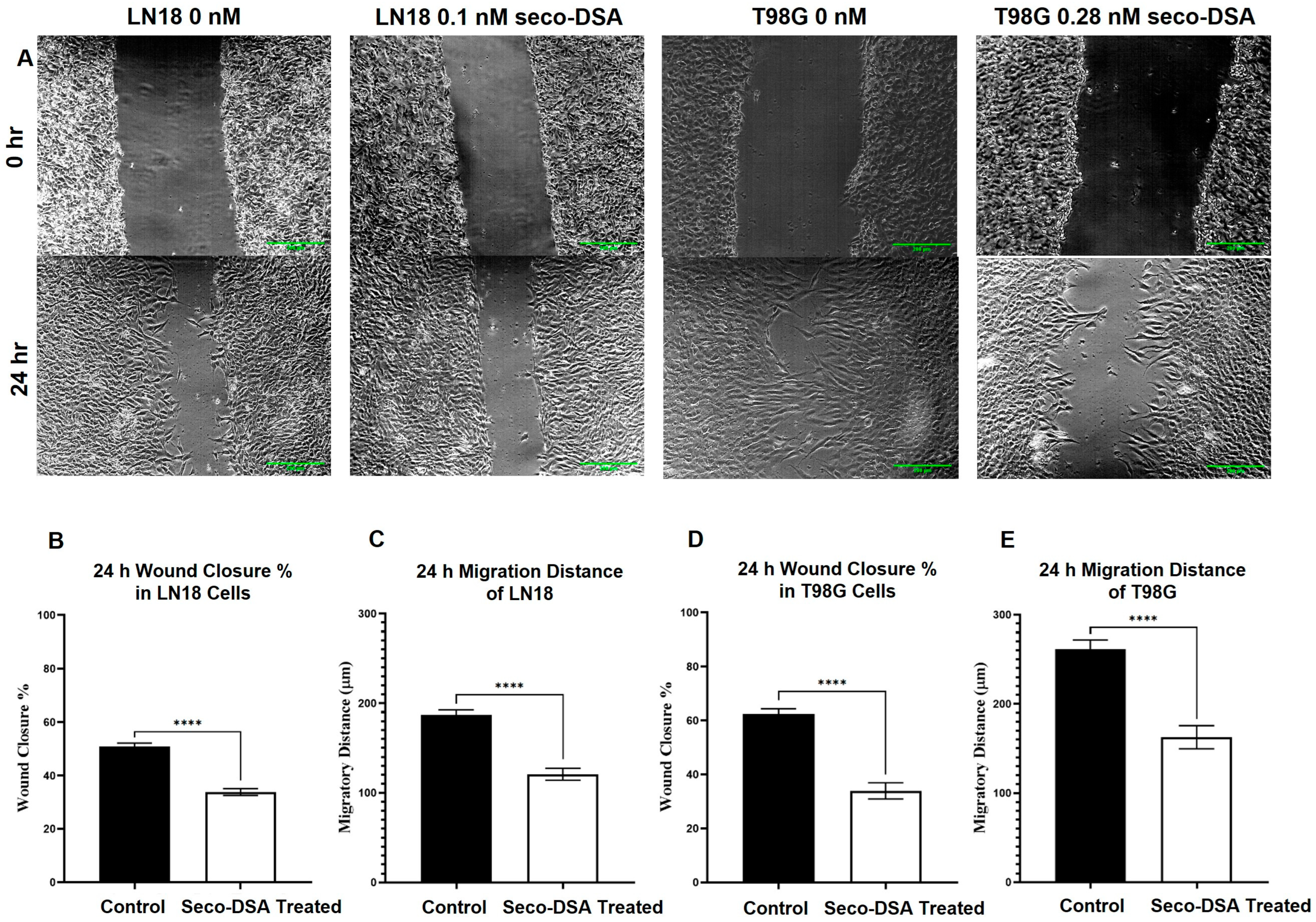
2.6. Cell Migration Is Inhibited by Seco-DSA
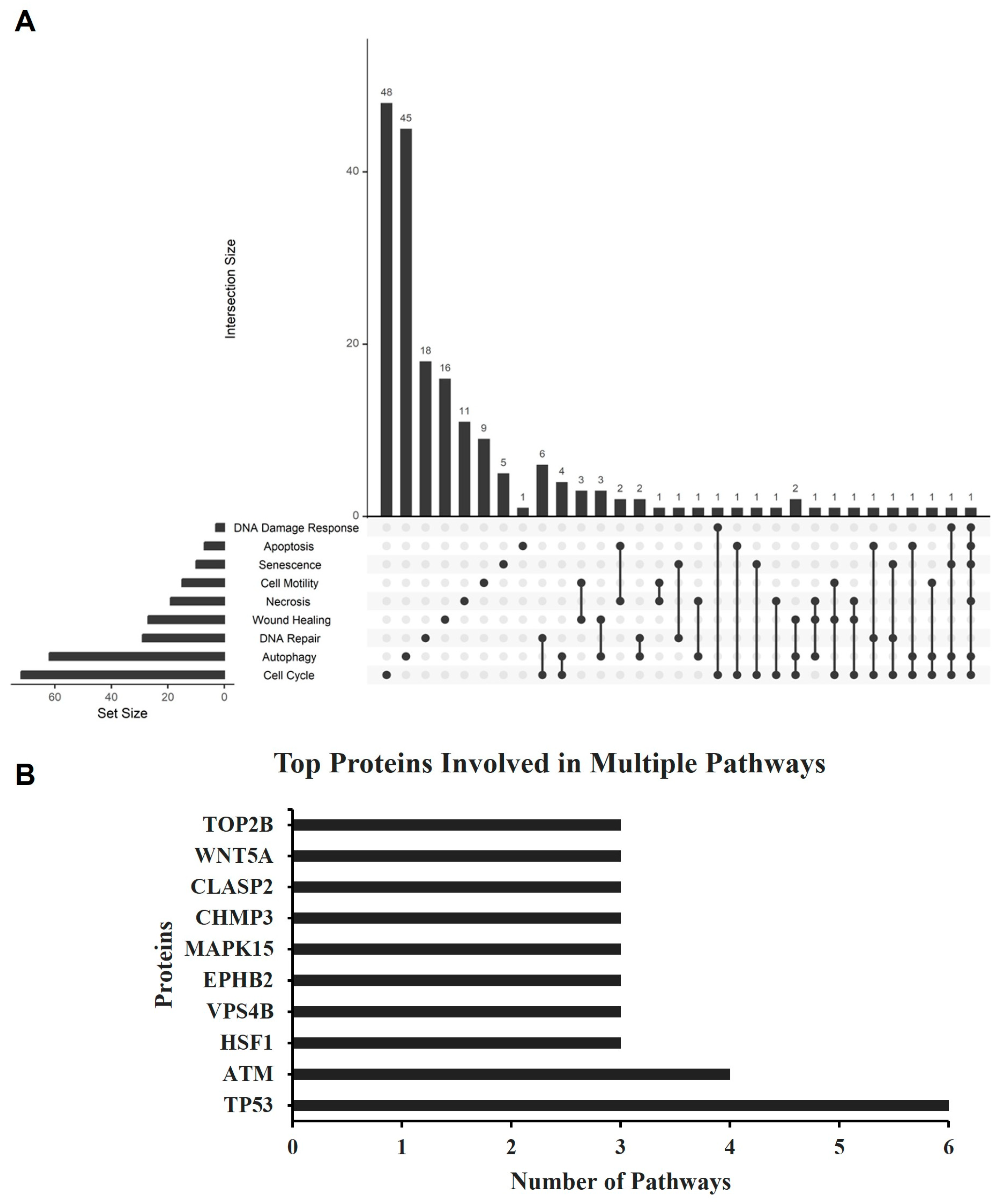
2.7. Proteomics Analysis
3. Discussion
4. Material and Methods
4.1. Cell Lines and Cell Culture
4.2. DSA Treatment
4.3. Colony Formation Assay
4.4. Cell Proliferation Assay
4.5. Methyl Thiazolyl Tetrazolium (MTT) Assay
4.6. Apoptosis and Necrosis Assay
4.7. Cell Cycle Analysis
4.8. Scratch Assay
4.9. Proteomic Analysis Sample Preparation
4.10. LC-MS/MS Data Acquisition
4.11. LC-MS/MS Data Search
4.12. Protein Mass Spec Analysis
4.13. Mass Spec Proteomics Data Accessibility
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iacob, G.; Dinca, E.B. Current data and strategy in glioblastoma multiforme. J. Med. Life 2009, 2, 386–393. [Google Scholar] [PubMed]
- Yesudhas, D.; Dharshini, S.A.P.; Taguchi, Y.H.; Gromiha, M.M. Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis. Genes 2022, 13, 428. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2020, 11, 603911. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef]
- Glas, M.; Rath, B.H.; Simon, M.; Reinartz, R.; Schramme, A.; Trageser, D.; Eisenreich, R.; Leinhaas, A.; Keller, M.; Schildhaus, H.U.; et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann. Neurol. 2010, 68, 264–269. [Google Scholar] [CrossRef]
- Ius, T.; Isola, M.; Budai, R.; Pauletto, G.; Tomasino, B.; Fadiga, L.; Skrap, M. Low-grade glioma surgery in eloquent areas: Volumetric analysis of extent of resection and its impact on overall survival. A single-institution experience in 190 patients: Clinical article. J. Neurosurg. 2012, 117, 1039–1052. [Google Scholar] [CrossRef]
- Arora, A.; Somasundaram, K. Glioblastoma vs temozolomide: Can the red queen race be won? Cancer Biol. Ther. 2019, 20, 1083–1090. [Google Scholar] [CrossRef]
- Fekete, B.W.K.; Tisell, M.; Pivodic, A.; Smits, A.; Jakola, A.S.; Rydenhag, B. What predicts survival in glioblastoma? A population-based study of changes in clinical management and outcome. Front. Surg. 2023, 10, 1249366. [Google Scholar] [CrossRef]
- Morcos, A.; Jung, Y.; Galvan Bustillos, J.; Fuller, R.N.; Caba Molina, D.; Bertucci, A.; Boyle, K.E.; Vazquez, M.E.; Wall, N.R. A Comprehensive Review of the Antitumor Properties and Mechanistic Insights of Duocarmycin Analogs. Cancers 2024, 16, 3293. [Google Scholar] [CrossRef] [PubMed]
- Eis, P.S.; Smith, J.A.; Rydzewski, J.M.; Case, D.A.; Boger, D.L.; Chazin, W.J. High resolution solution structure of a DNA duplex alkylated by the antitumor agent duocarmycin SA. J. Mol. Biol. 1997, 272, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.E.; Boger, D.L.; Wroe, A.; Vazquez, M. Duocarmycin SA, a potent antitumor antibiotic, sensitizes glioblastoma cells to proton radiation. Bioorg. Med. Chem. Lett. 2018, 28, 2688–2692. [Google Scholar] [CrossRef]
- Robertson, W.M.; Kastrinsky, D.B.; Hwang, I.; Boger, D.L. Synthesis and evaluation of a series of C5′-substituted duocarmycin SA analogs. Bioorg. Med. Chem. Lett. 2010, 20, 2722–2725. [Google Scholar] [CrossRef]
- Ichimura, M.; Ogawa, T.; Katsumata, S.; Takahashi, K.; Takahashi, I.; Nakano, H. Duocarmycins, new antitumor antibiotics produced by Streptomyces; producing organisms and improved production. J. Antibiot. 1991, 44, 1045–1053. [Google Scholar] [CrossRef]
- Tietze, L.F.; Haunert, F.; Feuerstein, T.; Herzig, T. A Concise and Efficient Synthesis of seco-Duocarmycin SA. Eur. J. Org. Chem. 2003, 562–566. [Google Scholar] [CrossRef]
- Vielhauer, G.A.; Swink, M.; Parelkar, N.K.; Lajiness, J.P.; Wolfe, A.L.; Boger, D. Evaluation of a reductively activated duocarmycin prodrug against murine and human solid cancers. Cancer Biol. Ther. 2013, 14, 527–536. [Google Scholar] [CrossRef]
- Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Predolini, F.; Pettinella, F.; Masserdotti, A.; Zammataro, L.; Altwerger, G.; Buza, N.; et al. SYD985, a Novel Duocarmycin-Based HER2-Targeting Antibody-Drug Conjugate, Shows Antitumor Activity in Uterine and Ovarian Carcinosarcoma with HER2/Neu Expression. Clin. Cancer Res. 2017, 23, 5836–5845. [Google Scholar] [CrossRef]
- Yao, H.P.Z.H.; Hudson, R.; Tong, X.M.; Wang, M.H. Duocarmycin-based antibody-drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: Pharmaceutical strategy and clinical progress. Drug Discov. Today 2021, 26, 1857–1874. [Google Scholar] [CrossRef]
- Parakh, S.; Nicolazzo, J.; Scott, A.M.; Gan, H.K. Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them? Front. Oncol. 2021, 11, 718590. [Google Scholar] [CrossRef]
- Li, C.; Qin, S.; Wen, Y.; Zhao, W.; Huang, Y.; Liu, J. Overcoming the blood-brain barrier: Exosomes as theranostic nanocarriers for precision neuroimaging. J. Control. Release 2022, 349, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Nguyen, J.; Jhaveri, N.; Rosenstein-Sisson, R.; Hofman, F.M.; Schonthal, A.H. A novel temozolomide analog, NEO212, with enhanced activity against MGMT-positive melanoma in vitro and in vivo. Cancer Lett. 2015, 358, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Harrabi, S.; Combs, S.E.; Brons, S.; Haberer, T.; Debus, J.; Weber, K.J. Temozolomide in combination with carbon ion or photon irradiation in glioblastoma multiforme cell lines—Does scheduling matter? Int. J. Radiat. Biol. 2013, 89, 692–697. [Google Scholar] [CrossRef]
- Jukes, Z.; Morais, G.R.; Loadman, P.M.; Pors, K. How can the potential of the duocarmycins be unlocked for cancer therapy? Drug Discov. Today 2021, 26, 577–584. [Google Scholar] [CrossRef]
- Forgie, B.N.; Prakash, R.; Goyeneche, A.A.; Telleria, C.M. Vitality, viability, long-term clonogenic survival, cytotoxicity, cytostasis and lethality: What do they mean when testing new investigational oncology drugs? Discov. Oncol. 2024, 15, 5. [Google Scholar] [CrossRef]
- Henriksson, E.; Kjellen, E.; Wahlberg, P.; Wennerberg, J.; Kjellstrom, J.H. Differences in estimates of cisplatin-induced cell kill in vitro between colorimetric and cell count/colony assays. In Vitro Cell. Dev. Biol. Anim. 2006, 42, 320–323. [Google Scholar] [CrossRef]
- Landry, M.; Nelson, D.; Choi, E.; DuRoss, A.; Sun, C. Development of a G2/M arrest high-throughput screening method identifies potent radiosensitizers. Transl. Oncol. 2022, 16, 101336. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Iriondo, M.N.; Etxaniz, A.; Varela, Y.R.; Ballesteros, U.; Lazaro, M.; Valle, M.; Fracchiolla, D.; Martens, S.; Montes, L.R.; Goni, F.M.; et al. Effect of ATG12-ATG5-ATG16L1 autophagy E3-like complex on the ability of LC3/GABARAP proteins to induce vesicle tethering and fusion. Cell. Mol. Life Sci. 2023, 80, 56. [Google Scholar] [CrossRef]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Charlton, D.E. The minimum Do for cell killing for alpha-particle emitters uniformly distributed in an extended medium. Radiat. Res. 1998, 149, 646–648. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 2 October 2024).
- Gehlenborg, N. UpSetR: A More Scalable Alternative to Venn and Euler Diagrams for Visualizing Intersecting Sets. 2019. Available online: https://CRAN.R-project.org/package=UpSetR (accessed on 15 October 2024).
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | Necrosis | Senescence | Autophagy | |||
|---|---|---|---|---|---|---|
| Up | TP53 | TP53 | TP53 | TP53 | ATG16L1 | IKBKG |
| BCL2L1 | AKAP12 | TOP2B | MET | ATP6V0A1 | EHMT2 | |
| ASAH1 | MME | RAB5A | VTI1A | WNK1 | ||
| TRADD | MAP2K4 | TRIM23 | SCOC | MAPK15 | ||
| INPP5K | ATG2A | EXOC7 | EPHB2 | |||
| EPHB2 | WDR45B | MTCL1 | ||||
| YTHDC2 | ACBD5 | ATP6V1D | ||||
| Down | HTRA2 | CASP4 | ATM | HTRA2 | CALCOCO2 | RNF5 |
| CDK5RAP3 | FADD | SMC5 | VPS37C | FKBP8 | FYCO1 | |
| HSF1 | GBA1 | HMGA2 | LGALS8 | WDR81 | MLST8 | |
| CASP4 | WNT5A | ERCC1 | HGS | RNF213 | ATG5 | |
| FADD | CHUK | HMGA1 | SPTLC2 | MARK2 | SH3BP4 | |
| BCL10 | ZNF277 | ATP6V1C1 | WDFY3 | WDR45 | ||
| GBP2 | ATP6V1E1 | MTMR14 | TRIM21 | |||
| TMSB4X | ARFIP2 | PIK3C3 | CDK5 | |||
| CD47 | RAB8A | SVIP | ATM | |||
| SMPD4 | UFM1 | PIP4K2C | VPS4B | |||
| NUB1 | FOXK1 | RMC1 | CHMP3 | |||
| HDAC2 | BNIP3 | SNF8 | SETD2 | |||
| FUNDC2 | SPG11 | TIGAR | ||||
| CHMP4A | SNX18 | GBA1 | ||||
| CHMP1B | ||||||
| Cell Cycle | DNA Damage | DNA Repair | Cell Motility | Wound Healing | ||
|---|---|---|---|---|---|---|
| Up | TP53 | PDGFRB | TP53 | POLE | MAPK15 | EPHB2 |
| EHMT2 | CCNA2 | NBN | SPIRE1 | WASL | RAB27A | |
| WNK1 | CCNH | DTX3L | SYNJ2BP | VAV2 | ||
| MAPK15 | PLK1 | TOP2B | STK26 | FERMT3 | ||
| CIT | RAD23A | HDAC3 | STARD13 | RAP2B | ||
| XRCC3 | KIF14 | POLD3 | RAP2B | |||
| SYF2 | KANK2 | SUPT20H | ||||
| POLE | DTX3L | WRAP53 | ||||
| SPIRE1 | TOP2B | CBX8 | ||||
| TOM1L2 | LCMT1 | |||||
| CENPS | NBN | |||||
| ANAPC13 | YTHDC2 | |||||
| TAOK3 | FAM83D | |||||
| Down | HTRA2 | MCM4 | ATM | HSF1 | CLASP2 | VPS4B |
| CDK5RAP3 | MLH1 | SETD2 | ADCY3 | CHMP3 | ||
| HSF1 | CDC34 | TIGAR | NEURL1 | CHMP1B | ||
| TRIM21 | UBE2A | RAD51 | ENG | FUNDC2 | ||
| CDK5 | CKS1B | SMARCC2 | RABGEF1 | CHMP4A | ||
| ATM | SRPK2 | BABAM2 | ZMYND8 | WNT5A | ||
| VPS4B | CDC20 | TADA3 | HDAC2 | CLASP2 | ||
| CHMP3 | DLG1 | SUPT7L | GNA13 | PAPSS2 | ||
| KIF3B | IK | NSD2 | MIA3 | GPX1 | ||
| ANKRD17 | TOP3A | YY1 | ITGA5 | |||
| PIAS1 | FOSL1 | POLE2 | YAP1 | |||
| RAD51 | CLASP2 | MORF4L2 | METAP1 | |||
| SMARCC2 | CUL3 | APLF | HPS6 | |||
| BABAM2 | GINS1 | MCRS1 | AJUBA | |||
| WNT5A | CDC73 | PNKP | VKORC1 | |||
| SMC5 | MYO16 | SENP3 | ARHGAP35 | |||
| UBE2E2 | GSPT1 | HPF1 | AK3 | |||
| TUBGCP2 | APPL2 | PPP4R2 | SLC7A11 | |||
| RIOK2 | TK1 | TERF2IP | HPSE | |||
| TIPIN | SMC2 | HMGA2 | F11R | |||
| NLE1 | GIGYF2 | GNA13 | ||||
| C9orf78 | CTDP1 | MIA3 | ||||
| TELO2 | MAD1L1 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morcos, A.; Jung, Y.; Fuller, R.N.; Bertucci, A.; Nguyen, A.; Zhang, Q.; Emge, T.; Boyle, K.E.; Wall, N.R.; Vazquez, M. Seco-Duocarmycin SA in Aggressive Glioblastoma Cell Lines. Int. J. Mol. Sci. 2025, 26, 2766. https://doi.org/10.3390/ijms26062766
Morcos A, Jung Y, Fuller RN, Bertucci A, Nguyen A, Zhang Q, Emge T, Boyle KE, Wall NR, Vazquez M. Seco-Duocarmycin SA in Aggressive Glioblastoma Cell Lines. International Journal of Molecular Sciences. 2025; 26(6):2766. https://doi.org/10.3390/ijms26062766
Chicago/Turabian StyleMorcos, Ann, Yeonkyu Jung, Ryan N. Fuller, Antonella Bertucci, Amy Nguyen, Quanqing Zhang, Tobias Emge, Kristopher E. Boyle, Nathan R. Wall, and Marcelo Vazquez. 2025. "Seco-Duocarmycin SA in Aggressive Glioblastoma Cell Lines" International Journal of Molecular Sciences 26, no. 6: 2766. https://doi.org/10.3390/ijms26062766
APA StyleMorcos, A., Jung, Y., Fuller, R. N., Bertucci, A., Nguyen, A., Zhang, Q., Emge, T., Boyle, K. E., Wall, N. R., & Vazquez, M. (2025). Seco-Duocarmycin SA in Aggressive Glioblastoma Cell Lines. International Journal of Molecular Sciences, 26(6), 2766. https://doi.org/10.3390/ijms26062766