
Impact of the Human Leukocyte Antigen Complex on Idiopathic Pulmonary Fibrosis Development and Progression in the Sardinian Population

 , , , , , , , , , and
, , , , , , , , , and
Abstract
1. Introduction
2. Results
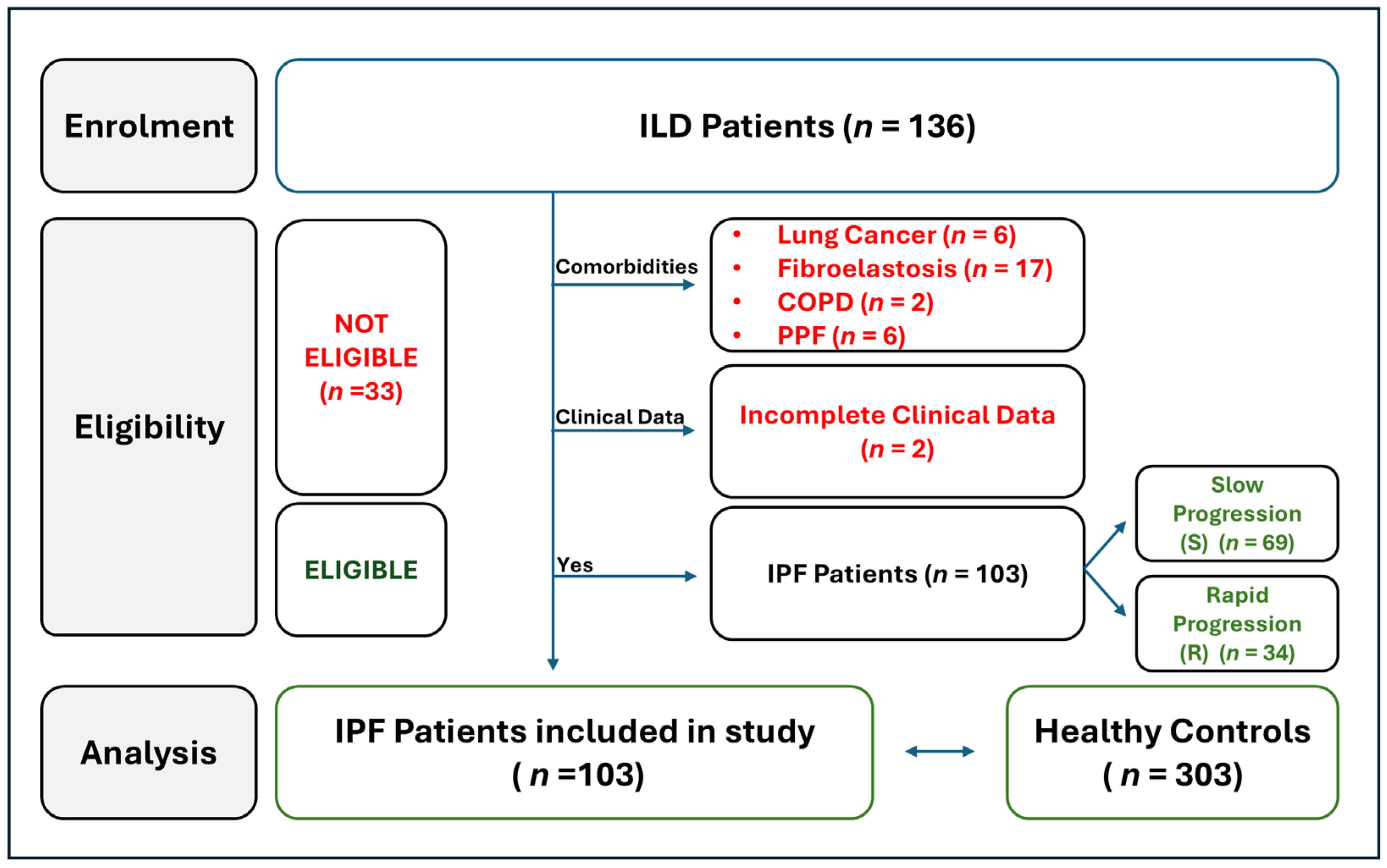
2.1. Workflow of IPF Patients Selection
2.2. Clinical and Demographic Baseline Parameters of IPF Patients
2.3. Comparison of HLA Allele and Haplotype Frequencies Between IPF Patients and Healthy Donors
2.4. Comparison of HLA Allele and Haplotype Frequencies Between IPF Patients Based on Disease Outcomes
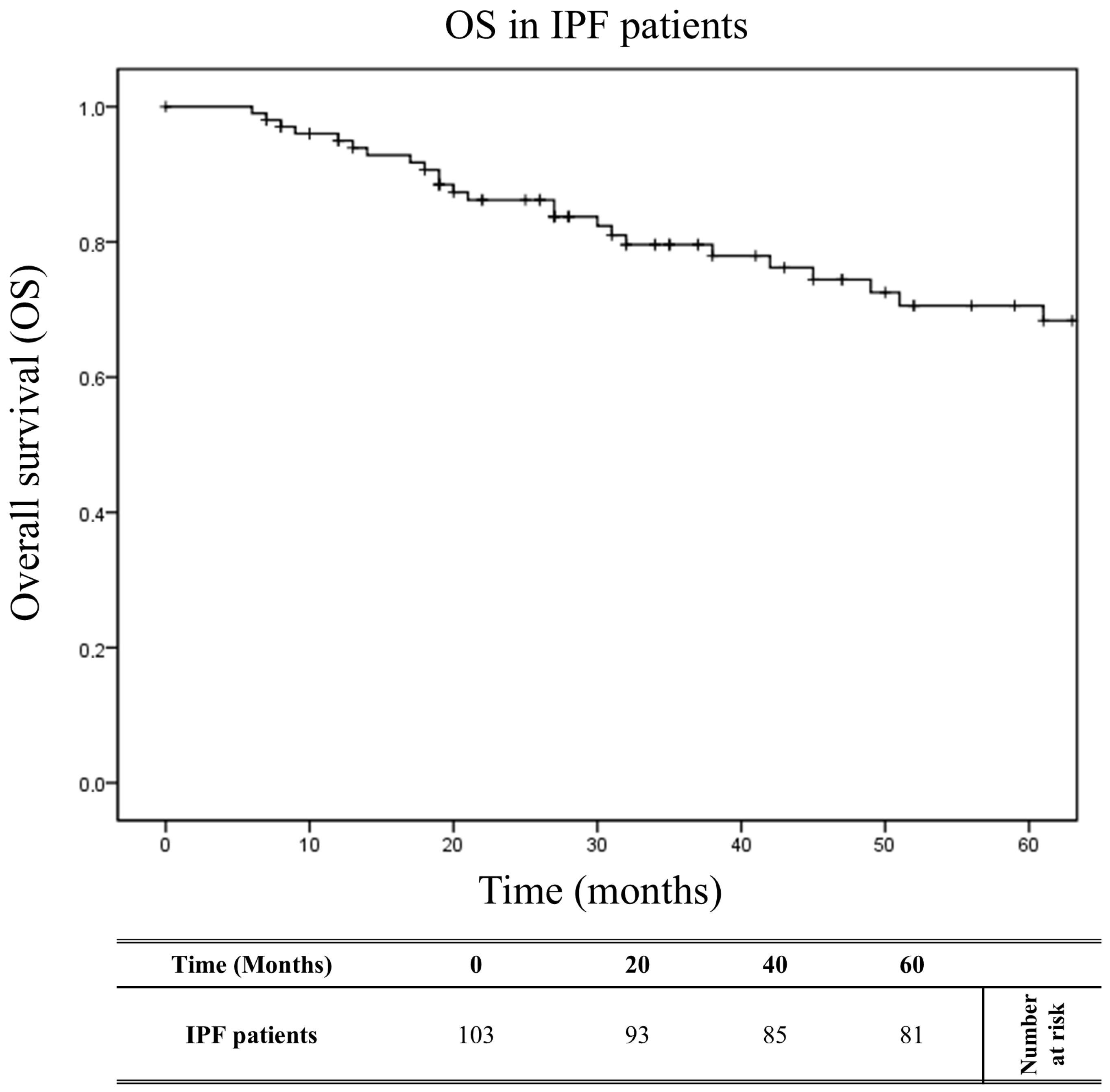
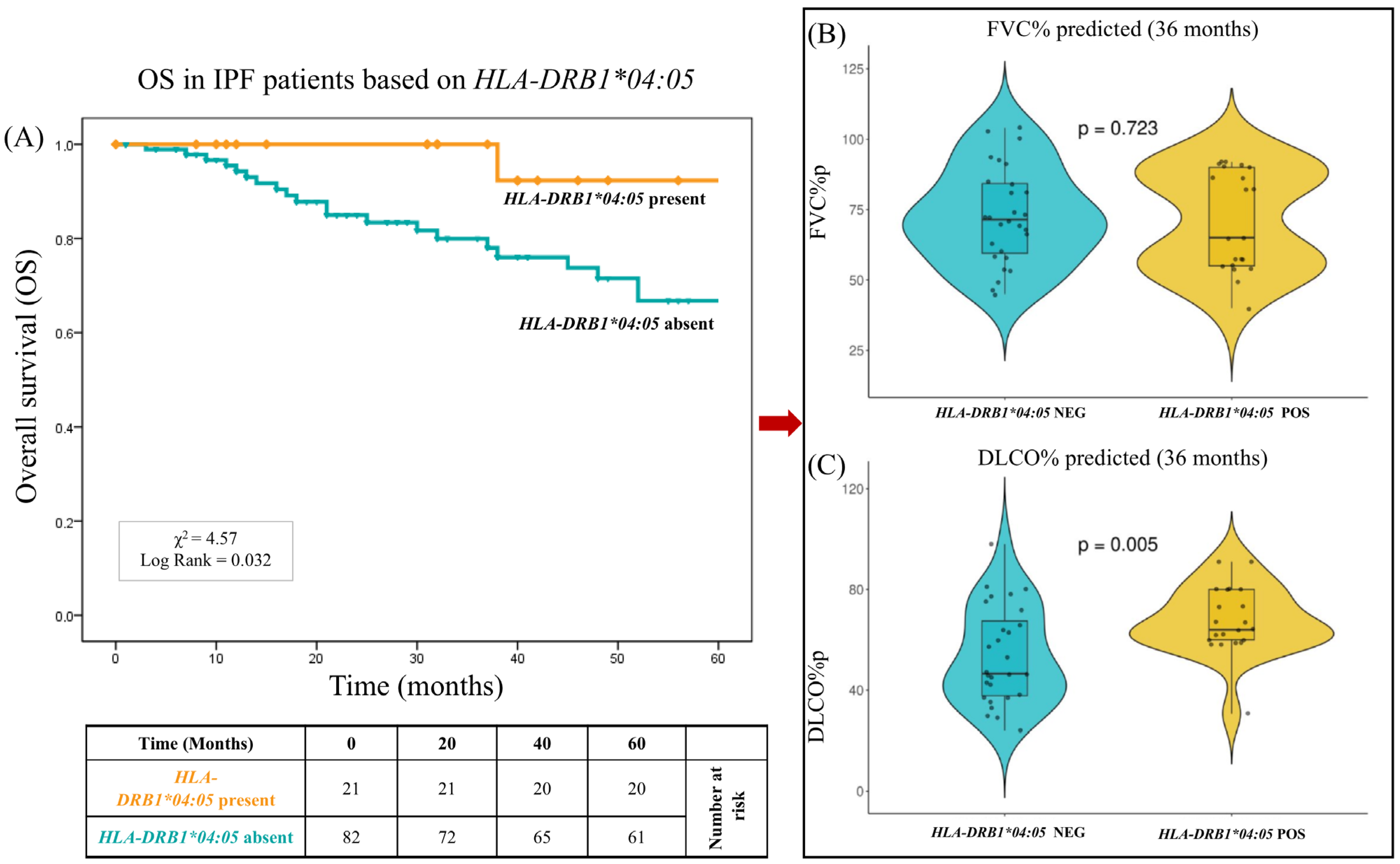
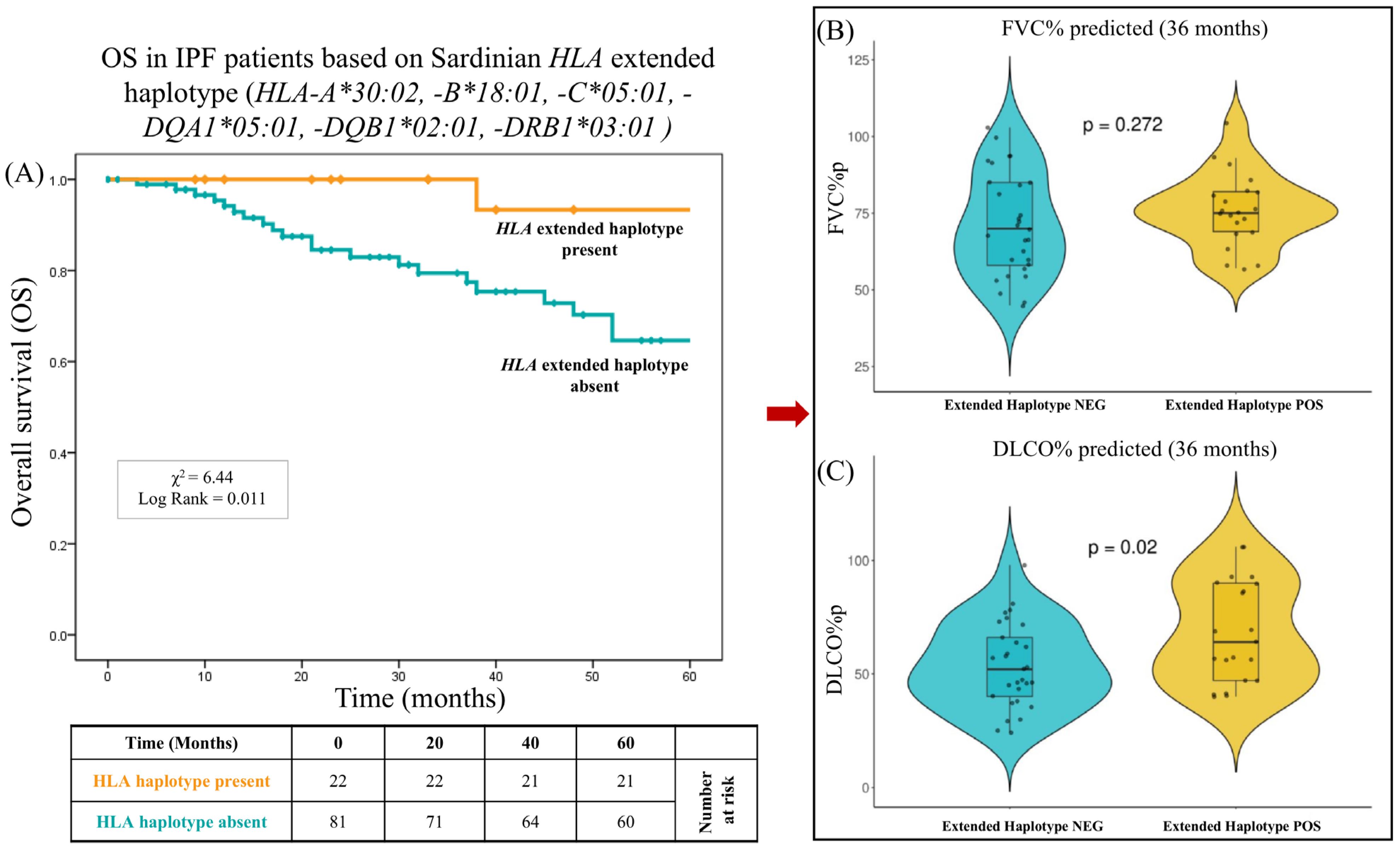
2.5. Impact of HLA on Overall Survival
3. Discussion
4. Materials and Methods
4.1. Study Cohorts
4.2. Ethics Statement
4.3. DNA Extraction and HLA Genotyping
4.4. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| AIH | Autoimmune hepatitis |
| COVID-19 | Coronavirus Disease 2019 |
| DILD | Drug-induced interstitial lung disease |
| DLCO | Carbon dioxide diffusing capacity of the lungs |
| HLA | Human leukocyte antigen |
| IPF | Idiopathic pulmonary fibrosis |
| LD | Linkage disequilibrium |
| RA | Rheumatoid arthritis |
| SE | Shared epitope |
| TB | Tuberculosis |
| TGF-β1 | Transforming growth factor-beta 1 |
References
- Raghu, G. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of fibrotic lung diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global Incidence and mortality of Idiopathic Pulmonary Fibrosis: A Systematic Review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Strongman, H.; Kausar, I.; Maher, T.M. Incidence, Prevalence, and Survival of Patients with Idiopathic Pulmonary Fibrosis in the UK. Adv. Ther. 2018, 35, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Raghu, G. Idiopathic Pulmonary Fibrosis in US Medicare Beneficiaries Aged 65 Years and Older: Incidence, Prevalence, and Survival, 2001–2011. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Hubbard, R. Occupational Dust Exposure and the Aetiology of Cryptogenic Fibrosing Alveolitis. Eur. Respir. J. Suppl. 2001, 32, 119–121. [Google Scholar] [CrossRef]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Stuart, B.D. Exome Sequencing Links Mutations in PARN and R℡1 with Familial Pulmonary Fibrosis and Telomere Shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA Variation and Disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Obeidat, M.; Hall, I.P. Genetics of Complex Respiratory Diseases: Implications for Pathophysiology and Pharmacology Studies. Br. J. Pharmacol. 2011, 163, 96–105. [Google Scholar] [CrossRef]
- Xue, J.; Gochuico, B.R.; Alawad, A.S.; Feghali-Bostwick, C.A.; Noth, I.; Nathan, S.D.; Rosen, G.D.; Rosas, I.O.; Dacic, S.; Ocak, I.; et al. The HLA Class II Allele DRB1*1501 Is Over-Represented in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2011, 6, e14715. [Google Scholar] [CrossRef]
- Guillen-Guio, B.; Paynton, M.L.; Allen, R.J.; Chin, D.P.W.; Donoghue, L.J.; Stockwell, A.; Leavy, O.C.; Hernandez-Beeftink, T.; Reynolds, C.; Cullinan, P. Association Study of Human Leukocyte Antigen Variants and Idiopathic Pulmonary Fibrosis. ERJ Open Res. 2024, 10, 00553–02023. [Google Scholar] [CrossRef]
- Olivieri, A.; Sidore, C.; Achilli, A.; Angius, A.; Posth, C.; Furtwängler, A.; Brandini, S.; Capodiferro, M.R.; Gandini, F.; Zoledziewska, M.; et al. Mitogenome Diversity in Sardinians: A Genetic Window onto an Island’s Past. Mol. Biol. Evol. 2017, 34, 1230–1239. [Google Scholar] [CrossRef]
- Di Gaetano, C.; Fiorito, G.; Ortu, M.F.; Rosa, F.; Guarrera, S.; Pardini, B.; Cusi, D.; Frau, F.; Barlassina, C.; Troffa, C.; et al. Sardinians Genetic Background Explained by Runs of Homozygosity and Genomic Regions under Positive Selection. PLoS ONE 2014, 9, e91237. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Hirschhorn, J.N. Small Island, Big Genetic Discoveries. Nat. Genet. 2015, 47, 1224–1225. [Google Scholar] [CrossRef] [PubMed]
- Contu, L.; Arras, M.; Carcassi, C.; Nasa, G.; Mulargia, M. HLA Structure of the Sardinian Population: A Haplotype Study of 551 Families. Tissue Antigens 1992, 40, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Fernández Fabrellas, E.; Peris Sánchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef]
- Littera, R.; Campagna, M.; Deidda, S.; Angioni, G.; Cipri, S.; Melis, M.; Firinu, D.; Santus, S.; Lai, A.; Porcella, R. Human Leukocyte Antigen Complex and Other Immunogenetic and Clinical Factors Influence Susceptibility or Protection to SARS-CoV-2 Infec-Tion and Severity of the Disease Course. Sardinian Exp. Front. Immunol. 2020, 11, 605688. [Google Scholar] [CrossRef]
- Chiang, C.W.K.; Marcus, J.H.; Sidore, C.; Biddanda, A.; Al-Asadi, H.; Zoledziewska, M.; Pitzalis, M.; Busonero, F.; Maschio, A.; Pistis, G. Genomic history of the Sardinian population. Nat. Genet. 2018, 50, 1426–1434. [Google Scholar] [CrossRef]
- Cilli, A.; Uzer, F. Disease Progression in Idiopathic Pulmonary Fibrosis under Anti-Fibrotic Treatment. Sarcoidosis Vasc. Diffus. Lung Dis. 2023, 40, 2023034. [Google Scholar]
- Lee, H.; Kim, S.Y.; Park, Y.S.; Choi, S.M.; Lee, J.H.; Park, J. Prognostic Implication of 1-Year Decline in Diffusing Capacity in Newly Di-Agnosed Idiopathic Pulmonary Fibrosis. Sci. Rep. 2024, 14, 8857. [Google Scholar]
- Umemura, T.; Katsuyama, Y.; Yoshizawa, K.; Kimura, T.; Joshita, S.; Komatsu, M.; Matsumoto, A.; Tanaka, E.; Ota, M. Human Leukocyte Antigen Class II Haplotypes Affect Clinical Characteristics and Progression of Type 1 Autoimmune Hepatitis in Japan. PLoS ONE 2014, 9, 100565. [Google Scholar] [CrossRef]
- Furumoto, Y.; Asano, T.; Sugita, T.; Abe, H.; Chuganji, Y.; Fujiki, K.; Sakata, A.; Aizawa, Y. Evaluation of the Role of HLA-DR Antigens in Japanese Type 1 Autoimmune Hepatitis. BMC Gastroenterol. 2015, 15, 144. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The Shared Epitope Hypothesis. An Approach to Understanding the Molecular Genetics of Susceptibility to Rheumatoid Arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Selvaraja, M.; Chin, V.K.; Abdullah, M.; Arip, M.; Amin-Nordin, S. HLA-DRB1*04 as a Risk Allele to Systemic Lupus Erythematosus and Lupus Nephritis in the Malay Population of Malaysia. Front. Med. 2020, 7, 598665. [Google Scholar] [CrossRef]
- Falfán-Valencia, R.; Camarena, A.; Pineda, C.L.; Montaño, M.; Juárez, A.; Buendía-Roldán, I.; Pérez-Rubio, G.; Reséndiz-Hernández, J.M.; Páramo, I.; Vega, A. Genetic Susceptibility to Multicase Hypersensitivity Pneumonitis Is Associated with the TNF-238 GG Genotype of the Promoter Region and HLA-DRB1*04 Bearing HLA Haplotypes. Respir. Med. 2014, 108, 211–217. [Google Scholar] [CrossRef]
- Imatoh, T.; Ushiki, A.; Ota, M.; Ito, M.; Sekine, A.; Yamashita, T.; Mashimo, Y.; Nakamura, R.; Saito, K.; Saito, Y. Association of HLA-DRB1*04:05 allele with drug-induced interstitial lung disease in Japanese population. Pharmacogenomics J. 2020, 20, 823–830. [Google Scholar] [CrossRef]
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782. [Google Scholar] [CrossRef]
- Azadeh, N.; Limper, A.H.; Carmona, E.M.; Ryu, J.H. The Role of Infection in Interstitial Lung Diseases: A Review. Chest 2017, 152, 842–852. [Google Scholar] [CrossRef]
- Taillé, C.; Grootenboer-Mignot, S.; Boursier, C.; Michel, L.; Debray, M.P.; Fagart, J.; Barrientos, L.; Mailleux, A.; Cigna, N.; Tubach, F. Identification of Periplakin as a New Target for Autoreactivity in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 759–766. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.A.; Tsai, C.G.; Valentine, V.G.; Kantrow, S.; Stoner, M.W.; Pilewski, J.M.; Gadgil, A.; George, M.P.; Gibson, K.F.; Choi, A.M. Cellular and Humoral Autoreactivity in Idiopathic Pulmonary Fibrosis. J. Immunol. 2007, 179, 2592–2599. [Google Scholar] [CrossRef]
- Kurosu, K.; Takiguchi, Y.; Okada, O.; Yumoto, N.; Sakao, S.; Tada, Y.; Kasahara, Y.; Tanabe, N.; Tatsumi, K.; Weiden, M. Identification of Annexin 1 as a Novel Autoantigen in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. J. Immunol. 2008, 181, 756–767. [Google Scholar] [CrossRef]
- Papiris, S.A.; Kollintza, A.; Karatza, M.; Manali, E.D.; Sotiropoulou, C.; Milic-Emili, J.; Roussos, C.; Daniil, Z. CD8+ T Lymphocytes in Bronchoalveolar Lavage in Idiopathic Pulmonary Fibrosis. J. Inflamm. 2007, 4, 14. [Google Scholar] [CrossRef]
- Kotsianidis, I. Global Impairment of CD4+CD25+FOXP3+ Regulatory T Cells in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 1121–1130. [Google Scholar] [CrossRef]
- Kallenberg, C.G.; Schilizzi, B.M.; Beaumont, F.; Leij, L.; Poppema, S.; The, T.H. Expression of Class II Major Histocompatibility Complex Antigens on Alveolar Epithelium in Interstitial Lung Disease: Relevance to Pathogenesis of Idiopathic Pulmonary Fibrosis. J. Clin. Pathol. 1987, 40, 725–733. [Google Scholar] [CrossRef]
- Rosas, I.O.; Ren, P.; Avila, N.A.; Chow, C.K.; Franks, T.J.; Travis, W.D.; McCoy, J.P.; May, R.M.; Wu, H.P.; Nguyen, D.M. Early Interstitial Lung Disease in Familial Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 698–705. [Google Scholar] [CrossRef]
- Osoegawa, K.; Mack, S.J.; Udell, J.; Noonan, D.A.; Ozanne, S.; Trachtenberg, E.; Prestegaard, M. HLA Haplotype Validator for Quality Assessments of HLA Typing. Hum. Immunol. 2016, 77, 273–282. [Google Scholar] [CrossRef]
- Carcassi, C.; Trucco, G.; Trucco, M.; Contu, L. A New HLA-DR2 Extended Haplotype Is Involved in Insulin-Dependent Diabetes Mellitus Susceptibility. Hum. Immunol. 1991, 31, 159–164. [Google Scholar] [CrossRef]
- Littera, R.; Chessa, L.; Onali, S.; Figorilli, F.; Lai, S.; Secci, L.; Nasa, G.; Caocci, G.; Arras, M.; Melis, M. Exploring the Role of Killer Cell Immunoglobulin-Like Receptors and Their HLA Class I Ligands in Autoimmune Hepatitis. PLoS ONE 2016, 11, 0146086. [Google Scholar] [CrossRef]
- Pastorino, R.; Menni, C.; Barca, M.; Foco, L.; Saddi, V.; Gazzaniga, G.; Ferrai, R.; Mascaretti, L.; Dudbridge, F.; Berzuini, C. Association between Protective and Deleterious HLA Alleles with Multiple Sclerosis in Central East Sardinia. PLoS ONE 2009, 4, 6526. [Google Scholar] [CrossRef]
- Bitti, P.P.; Murgia, B.S.; Ticca, A.; Ferrai, R.; Musu, L.; Piras, M.L.; Puledda, E.; Campo, S.; Durando, S.; Montomoli, C. Association between the Ancestral Haplotype HLA A30B18DR3 and Multiple Sclerosis in Central Sardinia. Genet. Epidemiol. 2001, 20, 271–283. [Google Scholar] [CrossRef]
- Contu, L.; Deschamps, I.; Lestradet, H.; Hors, J.; Schmid, M.; Busson, M.; Benajam, A.; Marcelli-Barge, A.; Dausset, J. HLA haplotype study of 53 juvenile insulin-dependent diabetic (I.D.D.) families. Tissue Antigens 1982, 20, 123–140. [Google Scholar] [CrossRef]
- Congia, M.; Frau, F.; Lampis, R.; Frau, R.; Mele, R.; Cucca, F.; Muntoni, F.; Porcu, S.; Boi, F.; Contu, L. A High Frequency of the A30, B18, DR3, DRw52, DQw2 Extended Haplotype in Sardinian Celiac Disease Patients: Further Evidence That Disease Susceptibility Is Conferred by DQ A1*0501, B1*0201. Tissue Antigens 1992, 39, 78–83. [Google Scholar] [CrossRef]
- Cambon-Thomsen, A.; Thomsen, M.; Abbal, M.; Sommer, E.; Calot, M.; Ohayon, E. A New HLA-D Specificity Associated with DR Blank: D-BON. Tissue Antigens 1986, 27, 256–261. [Google Scholar] [CrossRef]
- Contu, L.; Arras, M.; Mulargia, M.; Nasa, G.; Carcassi, C.; Leone, A.L.; Ledda, A.; Goddi, F. Study of HLA Segregation in 479 Tha-Lassemic Families. Tissue Antigens 1992, 39, 58–67. [Google Scholar] [CrossRef]
- Littera, R.; Perra, A.; Miglianti, M.; Piras, I.S.; Mocci, S.; Lai, S.; Melis, M.; Zolfino, T.; Balestrieri, C.; Conti, M. The double-sided of human leukocyte antigen-G molecules in type 1 autoimmune hepatitis. Front. Immunol. 2022, 13, 1007647. [Google Scholar] [CrossRef]
- Martelli-Palomino, G.; Pancotto, J.A.; Muniz, Y.C.; Mendes-Junior, C.T.; Castelli, E.C.; Massaro, J.D.; Krawice-Radanne, I.; Poras, I.; Reb-mann, V.; Carosella, E.D. Polymorphic Sites at the 3’ Untranslated Region of the HLA-G Gene Are Associated with Differential Hla-g Soluble Levels in the Brazilian and French Population. PLoS ONE 2013, 8, 71742. [Google Scholar] [CrossRef]
- Malkova, A.; Starshinova, A.; Zinchenko, Y.; Basantsova, N.; Mayevskaya, V.; Yablonskiy, P.; Shoenfeld, Y. The Opposite Effect of Human Leukocyte Antigen Genotypes in Sarcoidosis and Tuberculosis: A Narrative Review of the Literature. ERJ Open Res. 2020, 6, 00155–02020. [Google Scholar] [CrossRef]
- Wells, A.U.; Brown, K.K.; Flaherty, K.R.; Kolb, M.; Thannickal, V.J. What’s in a Name? That Which We Call IPF, by Any Other Name Would Act the Same. Eur. Respir. J. 2018, 51, 1800692. [Google Scholar] [CrossRef]
- Melis, M.; Littera, R.; Cocco, E.; Frau, J.; Lai, S.; Congeddu, E.; Ragatzu, P.; Serra, M.; Loi, V.; Maddi, R. Entropy of Human Leukocyte Antigen and Killer-Cell Immunoglobulin-like Receptor Systems in Immune-Mediated Disorders: A Pilot Study on Multiple Sclerosis. PLoS ONE 2019, 14, 0226615. [Google Scholar] [CrossRef]
- Tanha, K.; Mohammadi, N.; Janani, L. P-Value: What Is and What Is Not. Med. J. Islam. Repub. Iran 2017, 31, 65. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 17 January 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of Sardinian IPF Patients | Total IPF Pts n = 103 | R Group n = 34 | S Group n = 69 | Comparison of Group R vs. Group S | ||||
|---|---|---|---|---|---|---|---|---|
| Clinical parameters | Mean ± SD | Mean ± SD | Mean ± SD | p Value | † (95% CI) | |||
| Age at diagnosis (yr) | 70.0 ± 8.2 | 69.1 ± 8.3 | 70.0 ± 8.2 | 0.603 | −0.9 (−4.3; 2.5) | |||
| BMI | 26.4 ± 3.6 | 27.4 ± 4.1 | 26.1 ± 3.5 | 0.105 | 1.3 (−0.3; 2.8) | |||
| FVC%p, basal | 80.3 ± 18.5 | 70.4 ± 18.5 | 85.0 ± 18.7 | 0.0003 | −14.6 (−22.3; −6.9) | |||
| FVC%p, 2 yrs | 77.7 ± 20.5 | 66.3 ± 6.8 | 82.6 ± 22.8 | 0.0001 | −16.3 (−24.2; −8.4) | |||
| DLCO%p, basal | 63.1 ± 16.8 | 49.2 ± 16.8 | 69.6 ± 17.0 | <0.0001 | −20.4 (−27.4; −13.4) | |||
| DLCO%p, 2 yrs | 61.0 ± 19.6 | 36.7 ± 19.3 | 71.4 ± 15.4 | <0.0001 | −34.7 (−41.7; −27.7) | |||
| Demographic parameters | n | % | n | % | n | % | p Value | OR (95% CI) |
| O2 therapy | 33 | 0.320 | 26 | 0.765 | 7 | 0.101 | <0.0001 | 28.8 (9.5–87.6) |
| Age ≤ 65 yr | 15 | 0.146 | 5 | 0.147 | 10 | 0.145 | 1.000 | 1.0 (0.32–3.3) |
| Male | 79 | 0.767 | 27 | 0.794 | 52 | 0.755 | 0.805 | 1.3 (0.47–3.41) |
| Female | 24 | 0.233 | 7 | 0.206 | 17 | 0.246 | 0.805 | 0.7 (0.29–2.1) |
| Smoking history | 74 | 0.718 | 23 | 0.676 | 51 | 0.739 | 0.642 | 1.4 (0.55–3.3) |
| Antifibrotic Therapy | n | % | n | % | n | % | p Value | OR (95% CI) |
| Nintedanib | 59 | 0.573 | 20 | 0.588 | 39 | 0.565 | 1.000 | 1.1 (048–2.5) |
| Pirfenidone | 24 | 0.233 | 9 | 0.265 | 15 | 0.217 | 0.625 | 1.3 (0.50–3.4) |
| No compliance | 20 | 0.194 | 5 | 0.147 | 15 | 0.217 | 0.442 | 0.6 (0.21–1.9) |
| Overall Survival | n | % | n | % | n | % | p Value | OR (95% CI) |
| 12 months | 98 | 0.951 | 29 | 0.853 | 69 | 1.000 | 0.003 | 0.0 (0.0–0.5) |
| 36 months | 89 | 0.864 | 21 | 0.618 | 68 | 0.986 | <0.0001 | 42.1 (5.2–341) |
| 60 months | 82 | 0.796 | 16 | 0.471 | 66 | 0.951 | <0.0001 | 24.8 (6.5–94.4) |
| IPF Patients n = 103 | Controls n = 303 | |||||
|---|---|---|---|---|---|---|
| HLA Single Alleles | 2n = 206 | (%) | 2n = 606 | (%) | p | OR (95%CI) |
| Susceptibility | ||||||
| HLA-DQB1*04:01:01 | 3 | 1.46 | 0 | 0 | 0.016 | >1.22 |
| Protective | ||||||
| HLA-C*04:01:01 | 15 | 7.28 | 83 | 13.70 | 0.013 | 0.50 (0.26–0.89) |
| HLA-DPB1*04:02:01 | 9 | 4.37 | 79 | 13.04 | <0.0001 | 0.31 (0.13–0.62) |
| Two-loci HLA haplotypes | 2n = 206 | (%) | 2n = 606 | (%) | p | OR (95%CI) |
| Susceptibility | ||||||
| HLA-A*30:02:01, DQB1*02:02:01 | 5 | 2.43 | 2 | 0.33 | 0.013 | 7.51 (1.45–39.02) |
| HLA-A*32:01:01, DRB1*03:01:01 | 7 | 3.40 | 5 | 0.83 | 0.014 | 4.30 (1.35–13.68) |
| HLA-A*02:01:01, DRB1*04:05:01 | 5 | 2.43 | 1 | 0.17 | 0.005 | 15.05 (1.75–129.58) |
| HLA-A*32:01:01, HLA-C*02:02:02 | 6 | 2.91 | 2 | 0.33 | 0.004 | 9.06 (1.81–45.25) |
| HLA-C*02:02:02, DQA1*02:01:01 | 4 | 1.94 | 0 | 0 | 0.004 | >1.96 |
| Protective | ||||||
| HLA-A*11:01:01, HLA-C*04:01:01 | 2 | 0.97 | 28 | 4.62 | 0.017 | 0.20 (0.05–0.86) |
| HLA-C*04:01:01, DQB1*03:01:01 | 2 | 0.97 | 29 | 4.79 | 0.011 | 0.19 (0.04–0.82) |
| IPF R Group n = 34 | IPF S Group n = 69 | |||||
|---|---|---|---|---|---|---|
| 2n = 68 | (%) | 2n = 138 | (%) | p | OR (95% CI) | |
| HLA Alleles | ||||||
| Protective | ||||||
| - DRB1*04:05:01 | 2 | 2.94 | 20 | 14.49 | 0.014 | 0.18 (0.02–0.78) |
| Two-loci HLA haplotypes | ||||||
| Susceptibility | ||||||
| - A*01:01:01, DQB1*03:01:01 | 5 | 7.35 | 1 | 0.72 | 0.016 | 10.74 (1.17–516.5) |
| - A*02:01:01, DQB1*02:01:01 | 5 | 7.35 | 1 | 0.72 | 0.016 | 10.74 (1.17–516.5) |
| Protective | ||||||
| - B*49:01:01, HLA-C*07:01:01 | 0 | 0 | 12 | 8.70 | 0.010 | 0.13 (0.00–1.18) |
| Six Loci HLA Extended Haplotypes ^ | IPF R Group n = 34 | IPF S Group n = 69 | ||||
|---|---|---|---|---|---|---|
| 2n = 68 | (%) | 2n = 138 | (%) | p | OR (95%CI) | |
| HLA-A*30:02, B*18:01, C*05:01, DQA1*05:01, DQB1*02:01, DRB1*03:01 | 1 | 1.47 | 21 | 15.22 | 0.002 | 0.08 (0.01–0.63) |
| HLA-A*02:05, B*58:01, C*07:18, DQA1*01:02, DQB1*05:02, DRB1*16:01 | 4 | 5.88 | 9 | 6.52 | 1.000 | 0.90 (0.27–3.02) |
| HLA-A*02:01, B*18:01, C*05:01, DQA1*05:01, DQB1*02:01, DRB1*03:01 | 4 | 5.88 | 2 | 1.45 | 0.094 | 4.25 (0.76–23.8) |
| Characteristics of Sardinian IPF pts | Total Pts | IPF R Group | IPF S Group | Comparison of R Group vs. S Group | |||||
|---|---|---|---|---|---|---|---|---|---|
| (n = 103) | (%) | (n = 34) | (%) | (n = 69) | (%) | PU | PM | ORM (95% CIM) | |
| Age ≤ 55 yr | 6 | 0.058 | 1 | 0.167 | 5 | 0.833 | 0.661 | 0.766 | 1.48 (0.06–17.25) |
| Age ≥ 65 yr | 81 | 0.786 | 26 | 0.321 | 55 | 0.679 | 0.799 | 0.551 | 0.70 (0.21–2.34) |
| Male | 79 | 0.767 | 27 | 0.342 | 52 | 0.658 | 0.805 | 0.921 | 1.06 (0.33–3.58) |
| Smoking ^ | 74 | 0.718 | 25 | 0.338 | 49 | 0.662 | 1 | 0.643 | 0.77 (0.25–2.39) |
| IPF therapy | |||||||||
| Nintedanib | 59 | 0.573 | 20 | 0.339 | 39 | 0.661 | 1 | 0.832 | 0.90 (0.34–2.39) |
| Pirfenidone | 24 | 0.233 | 9 | 0.375 | 15 | 0.625 | 0.625 | 0.703 | 1.23 (0.41–3.65) |
| No therapy ° | 20 | 0.194 | 5 | 0.250 | 15 | 0.750 | 0.442 | 0.853 | 0.87 (0.19–3.56) |
| HLA alleles/haplotypes | (2n = 206) | (%) | (2n = 68) | (%) | (2n = 138) | (%) | |||
| - DRB1*04:05 | 22 | 0.107 | 2 | 0.029 | 20 | 0.145 | 0.014 | 0.010 | 0.11 (0.01–0.47) |
| - A*01:01, DQB1*03:01 | 6 | 0.029 | 5 | 0.074 | 1 | 0.007 | 0.016 | 0.121 | 1.62 (1.33–251.13) |
| - A*02:01, DQB1*02:01 | 6 | 0.029 | 5 | 0.074 | 1 | 0.007 | 0.016 | 0.121 | 1.62 (1.33–251.13) |
| - B*49:01, C*07:01 | 12 | 0.058 | 0 | 0 | 12 | 0.087 | 0.010 | 0.802 | 1.30 (0.14–11.01) |
| - A*30:02, B*18:01, C*05:01 | 31 | 0.150 | 3 | 0.097 | 28 | 0.903 | 0.001 | 0.164 | 0.26 (0.03–1.44) |
| - HLA-A*30:02, B*18:01, C*05:01, DQA1*05:01, DQB1*02:01, DRB1*03:01 | 22 | 0.107 | 1 | 0.045 | 21 | 0.955 | 7.9 × 10−4 | 0.010 | 0.065 (0.003–0.343) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, M.; Mocci, S.; Deidda, S.; Melis, M.; Chessa, L.; Lai, S.; Giuressi, E.; Mereu, C.; Sanna, C.; Lorrai, M.; et al. Impact of the Human Leukocyte Antigen Complex on Idiopathic Pulmonary Fibrosis Development and Progression in the Sardinian Population. Int. J. Mol. Sci. 2025, 26, 2760. https://doi.org/10.3390/ijms26062760
Serra M, Mocci S, Deidda S, Melis M, Chessa L, Lai S, Giuressi E, Mereu C, Sanna C, Lorrai M, et al. Impact of the Human Leukocyte Antigen Complex on Idiopathic Pulmonary Fibrosis Development and Progression in the Sardinian Population. International Journal of Molecular Sciences. 2025; 26(6):2760. https://doi.org/10.3390/ijms26062760
Chicago/Turabian StyleSerra, Marina, Stefano Mocci, Silvia Deidda, Maurizio Melis, Luchino Chessa, Sara Lai, Erika Giuressi, Caterina Mereu, Celeste Sanna, Michela Lorrai, and et al. 2025. "Impact of the Human Leukocyte Antigen Complex on Idiopathic Pulmonary Fibrosis Development and Progression in the Sardinian Population" International Journal of Molecular Sciences 26, no. 6: 2760. https://doi.org/10.3390/ijms26062760
APA StyleSerra, M., Mocci, S., Deidda, S., Melis, M., Chessa, L., Lai, S., Giuressi, E., Mereu, C., Sanna, C., Lorrai, M., Murgia, M., Cannas, F., Mascia, A., Perra, A., Littera, R., & Giglio, S. (2025). Impact of the Human Leukocyte Antigen Complex on Idiopathic Pulmonary Fibrosis Development and Progression in the Sardinian Population. International Journal of Molecular Sciences, 26(6), 2760. https://doi.org/10.3390/ijms26062760