1. Introduction
The liver is one of the largest and most vital organs in the human body, playing a central role in regulating energy metabolism by producing and storing essential nutrients [
1]. It performs multiple critical functions, such as detoxifying drugs and harmful substances, storing blood and various nutrients, and regulating the immune system [
2]. Due to its central role in metabolism and detoxification, the liver is particularly susceptible to damage from oxidative stress. Oxidative stress arises when there is an imbalance between the production of reactive oxygen species (ROS) and the body’s antioxidant defenses. Excessive ROS can damage key cellular components—proteins, lipids, and DNA—leading to impaired liver function [
3]. This oxidative damage is a common underlying mechanism in various liver diseases, including autoimmune, drug-induced, alcoholic, infectious, and congenital metabolic liver diseases [
4,
5,
6]. Patients suffering from oxidative liver damage may exhibit symptoms such as fatigue, anorexia, indigestion, jaundice, and a tendency to bleed, underscoring the critical impact of oxidative stress on liver health. ROS are involved in a range of physiological and pathological processes. In the liver, acute exposure to severe oxidative stress results in hepatocellular death and inflammation [
3]. ROS not only induce cellular damage but also influence gene expression in signal transduction pathways, affecting processes like cell growth and death [
7]. Given the significant role of oxidative stress in liver pathology, developing effective strategies to protect hepatocytes and prevent liver diseases is of paramount importance.
Natural products, especially flavonoids, have garnered significant attention for their potential to prevent oxidative liver damage [
8]. Flavonoids are a diverse group of plant secondary metabolites known for their antioxidant, anti-inflammatory, and hepatoprotective properties. They exert protective effects by scavenging free radicals, chelating metal ions, and modulating antioxidant enzyme activities, thereby mitigating oxidative stress and cellular damage in the liver [
9]. Among these flavonoids, isoquercitrin—a quercetin glycoside found in various medicinal herbs, fruits, and vegetables—has demonstrated a range of biological activities, including antioxidant, anti-inflammatory, and anticancer effects [
10,
11]. Isoquercitrin has been reported to reduce ROS levels, inhibit lipid peroxidation, and protect against oxidative stress-induced cellular damage [
12]. In particular, studies have shown that it induces apoptosis and autophagy in hepatocellular carcinoma cells via signaling pathways [
12]; exhibits antioxidant activity, including reduced ROS levels and the inhibition of in vivo and in vitro lipid peroxidation [
13]; and promotes neuroprotection and neurite elongation [
14]. However, the effects of isoquercitrin on oxidative liver injury and its underlying molecular mechanisms are not yet fully understood.
Recent advances have highlighted the importance of integrating in silico analyses with in vitro and in vivo experiments to fully understand the therapeutic potential of natural products [
15]. In silico methods, such as molecular docking, enable the prediction of interactions between bioactive compounds and target proteins, providing insights into their mechanisms of action [
16,
17]. For instance, kaempferol was shown to stimulate autophagy and regulate ferroptosis under oxidative stress through AMP-activated protein kinase (AMPK) signaling, with molecular docking confirming its binding to key proteins involved in these pathways [
18]. Similarly, an integrative approach combining in silico analysis and experimental validation was used to uncover the antioxidant properties of Bupleuri Radix and its active compounds [
19]. These studies demonstrate that combining computational modeling with experimental studies enhances our understanding of how natural products exert their biological effects.
In this study, we conducted a comprehensive investigation integrating in silico, in vitro, and in vivo experiments to evaluate the antioxidant and hepatoprotective effects of isoquercitrin against oxidative liver damage. We first observed that isoquercitrin exhibited a protective effect on hepatocytes subjected to oxidative stress induced by the arachidonic acid (AA) and iron model. Subsequently, we confirmed that isoquercitrin effectively inhibited ROS production in these cells. Molecular docking studies were performed to assess the stable binding affinity of isoquercitrin to key proteins within the AMP-activated protein kinase (AMPK) pathway, and we examined its impact on the activation of AMPK and acetyl-CoA carboxylase (ACC). Similar analyses were conducted on the Yes-associated protein (YAP) pathway to explore additional mechanisms underlying its hepatoprotective effects. Finally, we evaluated whether the oral administration of isoquercitrin could prevent liver toxicity using in vivo models. Through this integrated approach, our study aims to elucidate the potential of isoquercitrin as a therapeutic agent for protecting against oxidative liver injury.
3. Discussion
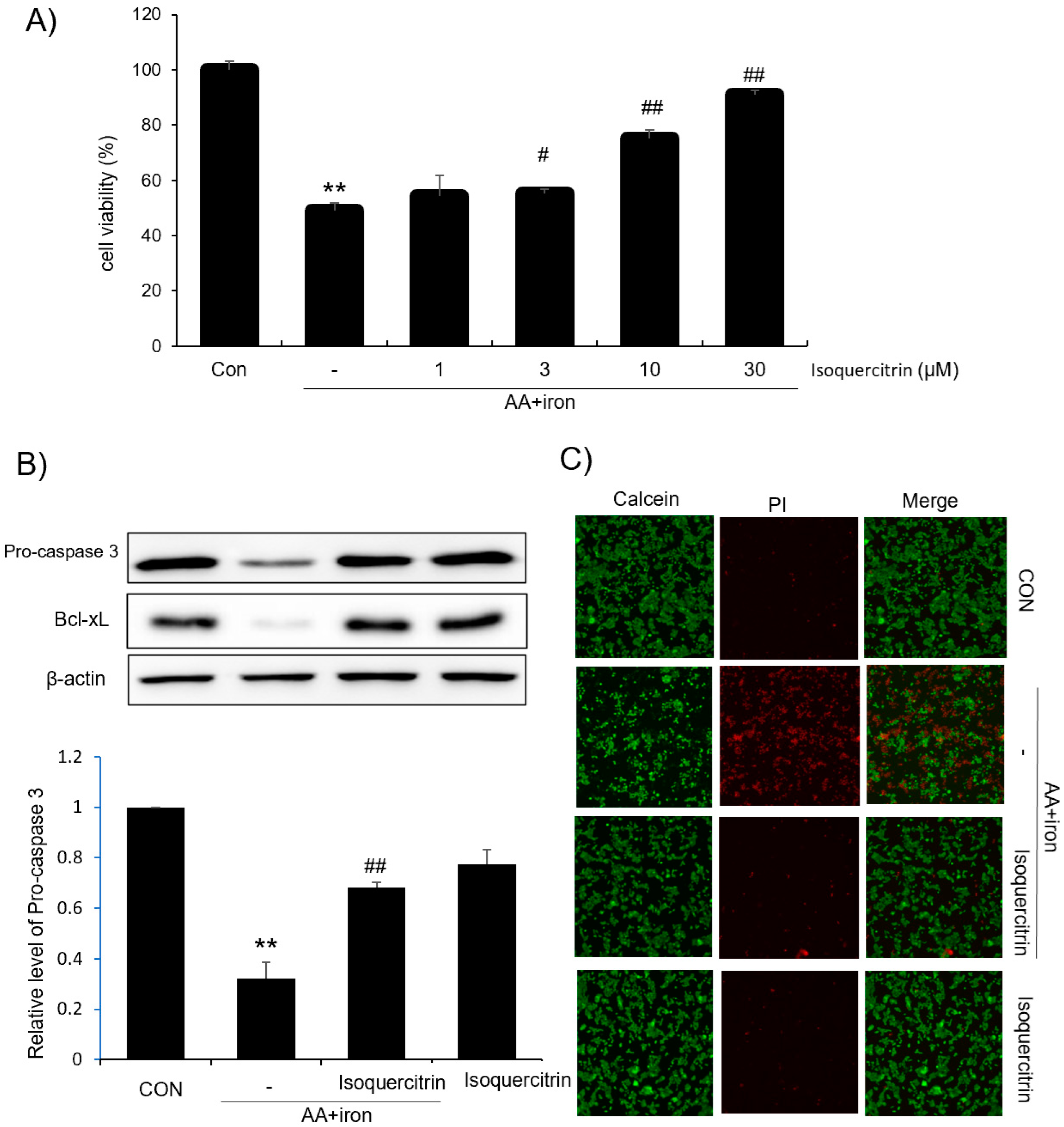
In this study, we evaluated the hepatoprotective and antioxidant effects of isoquercitrin and investigated its underlying molecular mechanisms. We found that isoquercitrin significantly enhanced cell viability in HepG2 cells exposed to AA+iron-induced oxidative stress (
Figure 1A). The cell viability increased in a dose-dependent manner, with the most effective protection observed at concentrations of 10 and 30 μg/mL. We also observed that isoquercitrin inhibited the decrease in apoptosis-related proteins Bcl-xL and procaspase-3 induced by AA+iron treatment (
Figure 1B), indicating its anti-apoptotic effects. Moreover, it is valuable to analyze the expression of the cleaved form of caspase 3 as well as procaspase-3 in the future. Fluorescence microscopy further confirmed that isoquercitrin reduced PI staining intensity compared to that in the AA+iron-treated group, suggesting that it prevented cell membrane damage and apoptosis (
Figure 1C). These findings suggest that isoquercitrin exerts cytoprotective effects by inhibiting apoptosis in hepatocytes under oxidative stress conditions. Also, we demonstrated that isoquercitrin effectively reduced ROS generation in a dose-dependent manner (
Figure 2A). The DCFH-DA assay showed that isoquercitrin significantly decreased ROS levels elevated by AA+iron treatment, demonstrating its potent antioxidant activity. Flow cytometry analysis revealed that isoquercitrin prevented the collapse of mitochondrial membrane potential induced by oxidative stress (
Figure 2B). By preserving mitochondrial function, isoquercitrin helps maintain cellular energy metabolism and prevent cell death.
A key finding was the activation of the AMPK pathway by isoquercitrin. Molecular docking analysis indicated strong binding affinities between isoquercitrin and AMPK-related proteins, such as AMPK, CaMKK2, ACC, and LKB1 (
Figure 3A). This suggests that isoquercitrin may directly interact with these proteins to modulate their activity. Western blot analysis showed that isoquercitrin increased the phosphorylation levels of AMPK and its downstream target ACC in a time-dependent manner, peaking at 3 h post-treatment (
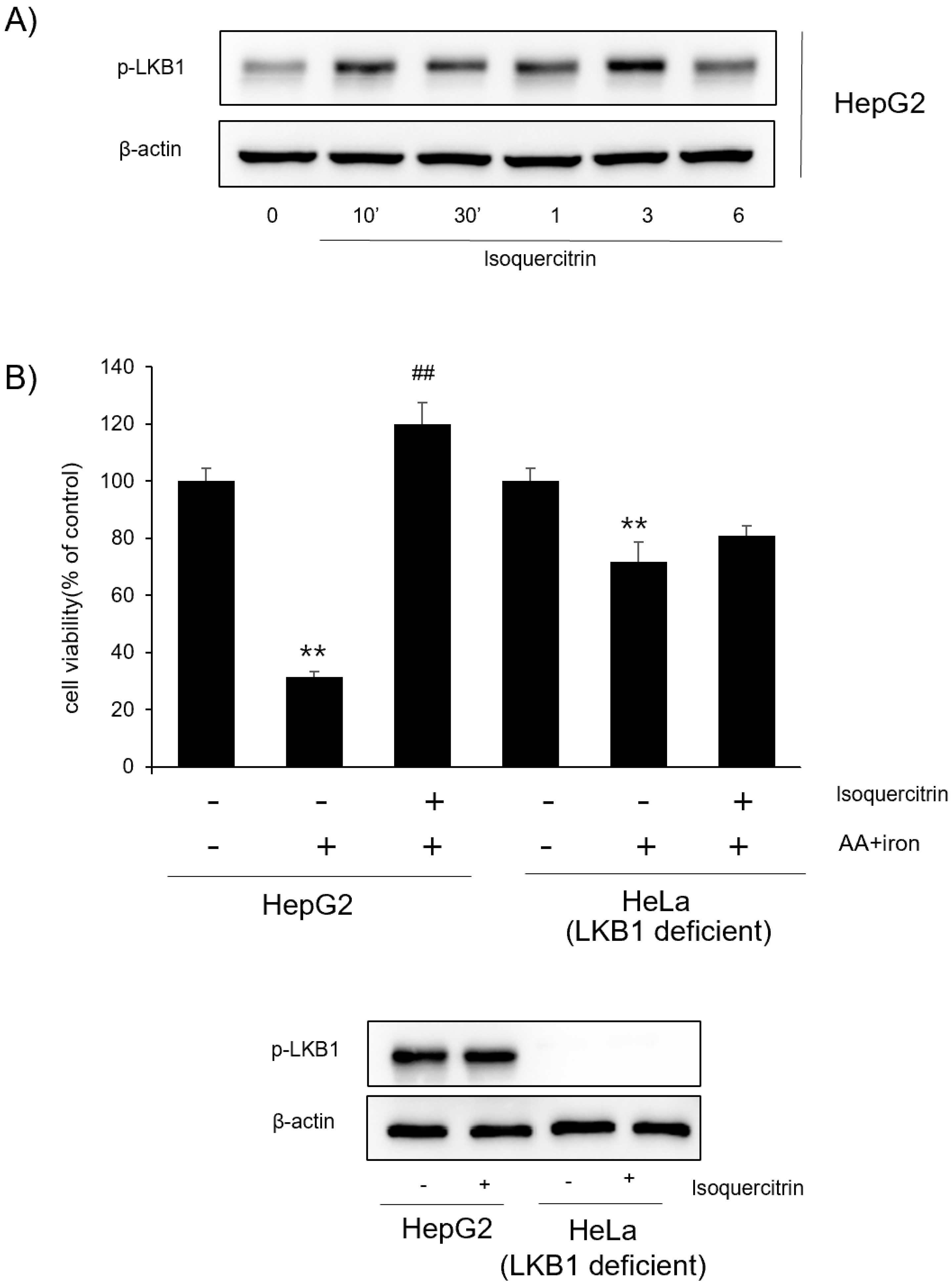
Figure 3B). This activation was consistent in both HepG2 and Huh7 cells, indicating a broader applicability. We further explored the role of LKB1 in isoquercitrin-induced AMPK activation. Our results showed that isoquercitrin increased the phosphorylation of LKB1 (
Figure 4A), suggesting that LKB1 is involved in the upstream regulation of AMPK activation by isoquercitrin. In LKB1-deficient HeLa cells, isoquercitrin failed to induce AMPK phosphorylation (
Figure 4B), confirming that LKB1 is essential for this pathway. These findings align with previous studies indicating that LKB1 is a primary activator of AMPK [
20,
21]. Activation of the LKB1/AMPK pathway by isoquercitrin has significant implications for energy metabolism. AMPK activation inhibits ACC, reducing fatty acid synthesis and promoting fatty acid oxidation [
22]. By regulating these metabolic enzymes, isoquercitrin helps restore energy homeostasis and may prevent metabolic disorders [
12,
23]. In fact, the molecular manipulation of AMPK in the liver cells is important in this study. Therefore, some molecular tools such as siRNA would be recommended in the future for similar studies with HeLa cells. Our study supports the notion that isoquercitrin can improve energy metabolism by activating the LKB1/AMPK pathway.
In addition to our findings, several independent studies have reported that isoquercitrin can activate or modulate the AMPK signaling pathway in diverse cellular contexts. For instance, a research demonstrated that isoquercitrin triggers apoptosis in bladder cancer cells via activation of the AMPK pathway, suggesting that isoquercitrin’s effects on energy metabolism are not restricted to hepatocytes [
24]. Similarly, isoquercitrin has been shown to induce both apoptosis and autophagy in hepatocellular carcinoma cells by modulating the AMPK/mTOR/p70S6K signaling axis, further supporting its role as an AMPK activator [
12]. Moreover, recent evidence indicates that isoquercitrin may enhance oxidative stress and promote ferroptosis in nasopharyngeal carcinoma cells via modulation of AMPK/NF-κB signaling [
11]. Collectively, these reports corroborate our observation that isoquercitrin activates the AMPK pathway and suggest that the compound’s beneficial effects on lipid metabolism and cell survival may be mediated, at least in part, by its modulation of AMPK signaling.
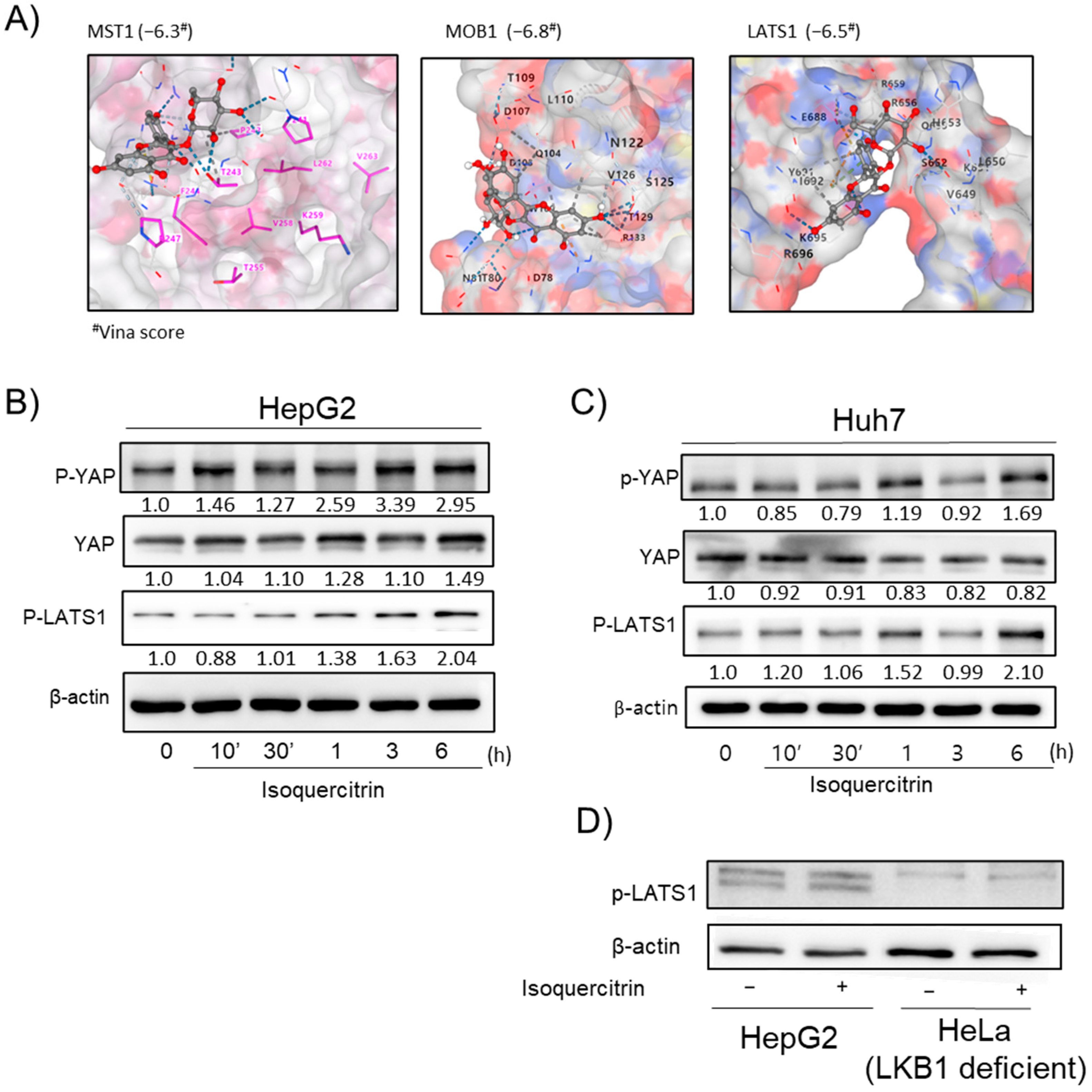
We also investigated the modulation of the YAP signaling pathway by isoquercitrin. Molecular docking suggested potential interactions between isoquercitrin and YAP pathway proteins, such as MST1, MOB1, and LATS1 (
Figure 5A). Western blot analysis showed that isoquercitrin enhanced YAP activity in HepG2 and Huh7 cells (
Figure 5B,C), but not in HeLa cells, indicating a cell-specific effect. The modulation of the YAP pathway may contribute to isoquercitrin’s hepatoprotective effects by influencing cell proliferation and survival pathways.
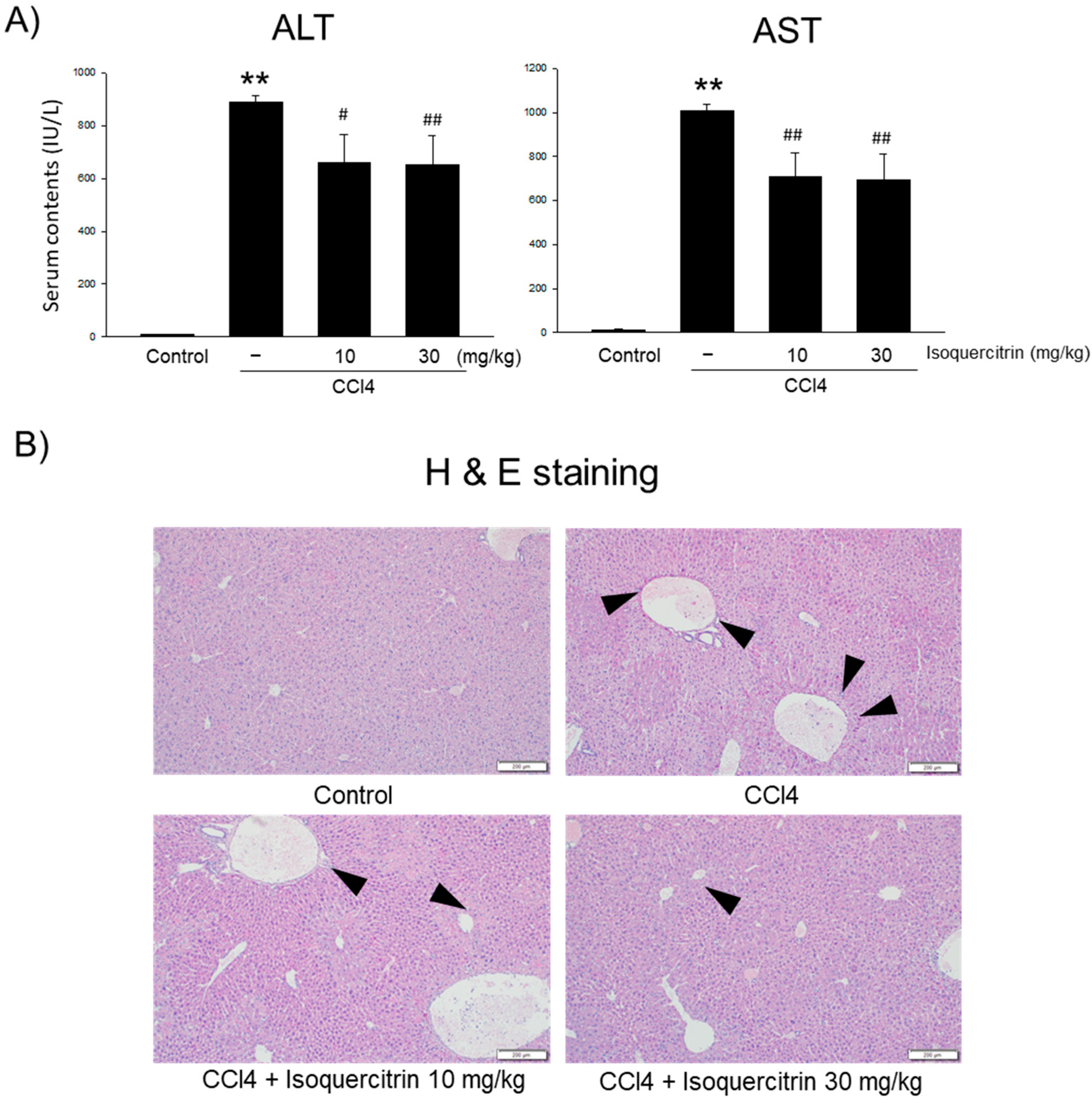
In our in vivo studies, we evaluated the protective effects of isoquercitrin on CCl
4-induced liver injury in mice. The oral administration of isoquercitrin significantly reduced serum ALT and AST levels compared to those in the CCl
4-only group (
Figure 6A), indicating decreased liver damage. Histopathological analysis confirmed that isoquercitrin mitigated liver tissue damage, preserving hepatic architecture and reducing necrosis and inflammation (
Figure 6B). Although the AMPK level in the liver in important, we could not confirm changes in protein expression in the liver, which need to be clarified in the future. These findings demonstrate that isoquercitrin’s hepatoprotective effects observed in vitro translate to in vivo models.
We employed a comprehensive strategy integrating molecular docking analyses with both in vitro and in vivo experiments to explore the effects of isoquercitrin. However, our study is not without limitations: Although our in vitro data and molecular docking analysis indicate a direct interaction between isoquercitrin and AMPK-related proteins, further validation using direct binding assays and primary hepatocyte models is needed. In future studies, it will be interesting to (1) perform in vivo experiments using NAFLD animal models to evaluate the long-term effects of isoquercitrin on hepatic lipid metabolism and (2) explore additional molecular mechanisms—such as potential crosstalk with mTOR and YAP signaling pathways—that may further explain the cytoprotective properties of isoquercitrin. These efforts will help to clarify the detailed molecular interactions underlying isoquercitrin-mediated AMPK activation and strengthen its potential application in the prevention of metabolic disorders.
4. Materials and Methods
4.1. Reagents
Isoquercitrin was purchased from Sigma-Aldrich. Some antibodies against anti-phospho AMP-activated protein kinase (p-AMPK, #2535), anti-AMPK, anti-phospho acetyl-CoA carboxylase (p-ACC, #3661), anti-poly (ADP-ribose) polymerase (PARP, #9542), anti-procaspase 3, anti-phospho liver kinase B1 (p-LKB1, #3482), liver kinase B1 (LKB1, #3050), and Bcl-xL were purchased from Cell Signaling Technology (Beverly, MA, USA), and anti-mouse and anti-rabbit antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Additionally, arachidonic acid (AA) and rhodamine123 (Rho123) were purchased from Calbiochem (San Diego, CA, USA), and 2′,7′-dichlorodihydrofluorescin diacetate (H2DCF-DA), ferric nitrilotriacetic acid (Fe-NTA), and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoleum bromide (MTT) were purchased from Sigma (St. Louis, MO, USA).
4.2. Cell Culture and Treatment
HepG2 cells, a human-derived liver parenchymal cell line, used in the experiment, as well as Hela cells and Huh7 cells, were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). All cells were cultured at 37 °C in a medium containing 10% bovine fetal serum (FBS), 100 μg/mL penicillin, and 1 mL Normocin in Dulbecco’s modified Eagle’s medium (DMEM) and 5% CO
2 in an incubator [
19].
Afterward, the cells were dispensed onto plates suitable for each experiment and incubated for 12 h with the above DMEM, which was then replaced with FBS-free and serum-deficient media for 12 h. Then, 1–30 μg/mL of isoquercitrin and 10 μM AA were added simultaneously for 12 h, followed by 5 μM iron for 1–6 h. All cells in the experiment were tested at 80–90% confluence.
4.3. Cell Viability Test (MTT Assay)
HepG2 cells were seeded in a 48-well plate at 1 × 10
5 cells/well and cultured for 24 h. After treatment with 0, 1, 3, 10, 30, and 100 µg/mL of isoquercitrin and 10 µM AA for 12 h, 5 µM iron was further added for 3 h. 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoleum bromide (MTT) reagent was added at 0.1 mg/mL and reacted at 37 °C for 1 h. The generated formazan crystals were dissolved with dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm using a microplate measuring device (Tecan Infinite
® M200 PRO, Baldwin Park, CA, USA) [
19].
4.4. Immunoblot Analysis
Cell lysates were prepared using lysis buffer (Thermo, Rockford, IL, USA), and were also homogenized with a sonicator (Sonics, USA). After centrifugation at 15,000×
g for 30 min, only the supernatant was collected, and the protein was quantified using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Next, the proteins extracted from the cells were separated by size by 10% SDS-PAGE and then electrotransferred from the acrylamide gel to a nitrocellulose membrane. Afterward, the nitrocellulose membrane was treated overnight at 4 °C with antibodies appropriate for each protein. HRP-linked anti-rabbit or anti-mouse secondary antibodies were used, and color was developed using ECL chemiluminescence detection reagents and then detected using a Chemidoc image analyzer (Vilber Lourmat, France) previously described [
19].
4.5. Measurement of Intracellular ROS
To measure the intracellular antioxidant activity of isoquercitrin, ROS was quantified using 2′,7′-dichlorofluorescein diacetate (DCFH-DA). HepG2 cells were treated with 10 µM DCFH-DA for 1 h at 37 °C to induce cell response, which was then oxidized to 2′,7′-dichlorofluorescein (DCF) by ROS and exhibited high fluorescence. The fluorescence level of DCF was analyzed using an ELISA microplate reader [
19].
4.6. Measurement of Mitochondrial Membrane Permeability (MMP)
To observe the mitochondrial protective effect of isoquercitrin, MMP was evaluated using flow cytometry after staining with rhodamine 123 (Rh123), a mitochondrion-specific green fluorescent dye. HepG2 cells were seeded at a density of 1 × 10
5 per well in 6-well plates for 24 h and then cultured in serum-free MEM for 12 h. After that, 30 µg/mL of isoquercitrin and 10 µM AA were added and reacted for 12 h, followed by 5 µM iron for 3 h and staining with 0.05 µg/mL Rh123 for 1 h. Cells were harvested using trypsin and resuspended in PBS, and MMP changes were measured using a flow cytometer (FACS, Partec, Münster, Germany). MMP was observed by recording a population of 10,000 cells [
19].
4.7. Fluorescence Microscopy
HepG2 cells were cultured in 6-well plates at 1 × 106 cells/well. Isoquercitrin 30 µg/mL was added with 10 µM AA for 12 h, and then 5 µM iron was reacted for 3 h. Then, calcein and PI were added at a concentration of 0.5 µM for 1 h.
4.8. Molecular Docking
Molecular docking studies were performed to predict the potential binding of isoquercitrin to key regulatory proteins in the AMPK and YAP signaling pathways using the CB-Dock2 online docking server (
https://cadd.labshare.cn/cb-dock2, accessed on 8 July 2024) [
25]. The three-dimensional structure of isoquercitrin (PubChem CID: 5280804) was obtained from the PubChem database in SDF format and directly uploaded to the CB-Dock2 server without further manual preprocessing. The crystal structures of the target proteins—CaMKK2 (PDB ID: 5UYJ), ACC (3H0J), AMPK (4CFE), LKB1 (2WTK), MST1 (3COM), MOB1 (1PI1), and LATS1 (5B5W)—were downloaded from the Protein Data Bank (PDB) in PDB format. For each docking run, the ligand (isoquercitrin) and protein structures were uploaded to the CB-Dock2 server, which automatically detects the binding pockets and performs docking calculations using default parameters. The server outputs a list of binding poses ranked by predicted binding energies (docking scores). The top-ranking poses were selected for subsequent visualization and analysis.
4.9. Statistical Treatment
The results of this study were expressed as the mean ± standard error (mean ± S.E.M.) of the results of experiments repeated at least three times. The comparison of each treatment group was performed using one-way analysis of variance (ANOVA) or Student’s t-test was used to verify statistical significance (p < 0.05 or p < 0.01)


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}