
Genomic Prediction for Germplasm Improvement Through Inter-Heterotic-Group Line Crossing in Maize



Abstract
1. Introduction
2. Results
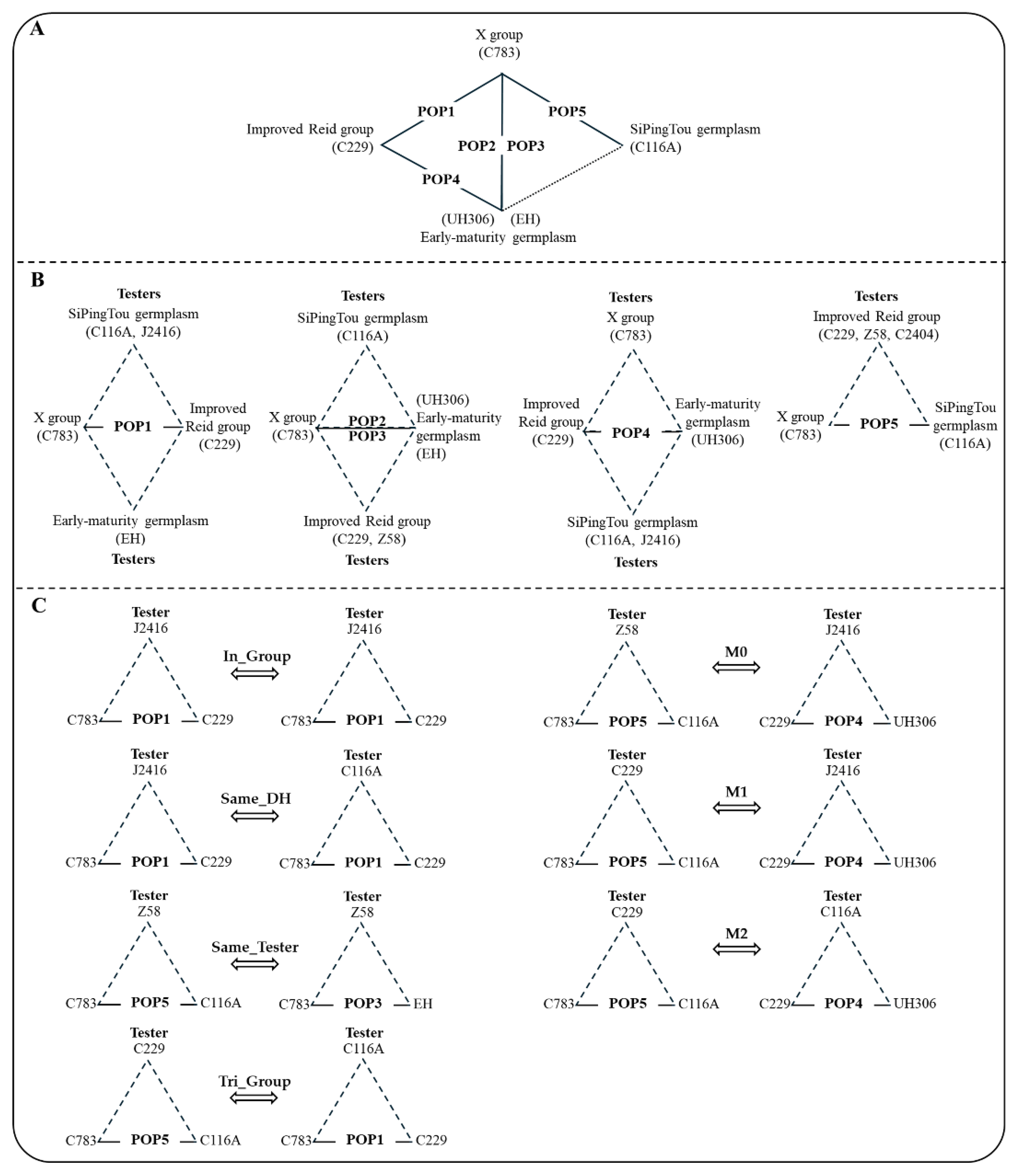
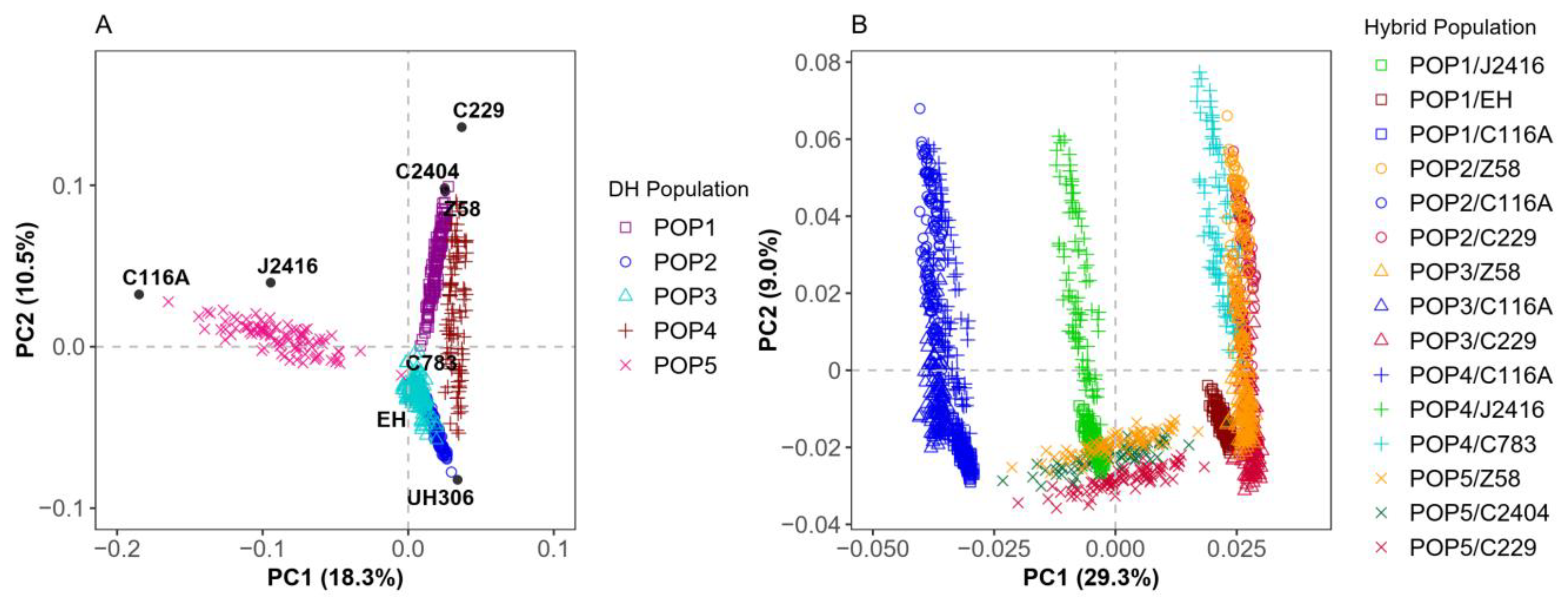
2.1. Genotyping and Population Structure Analysis
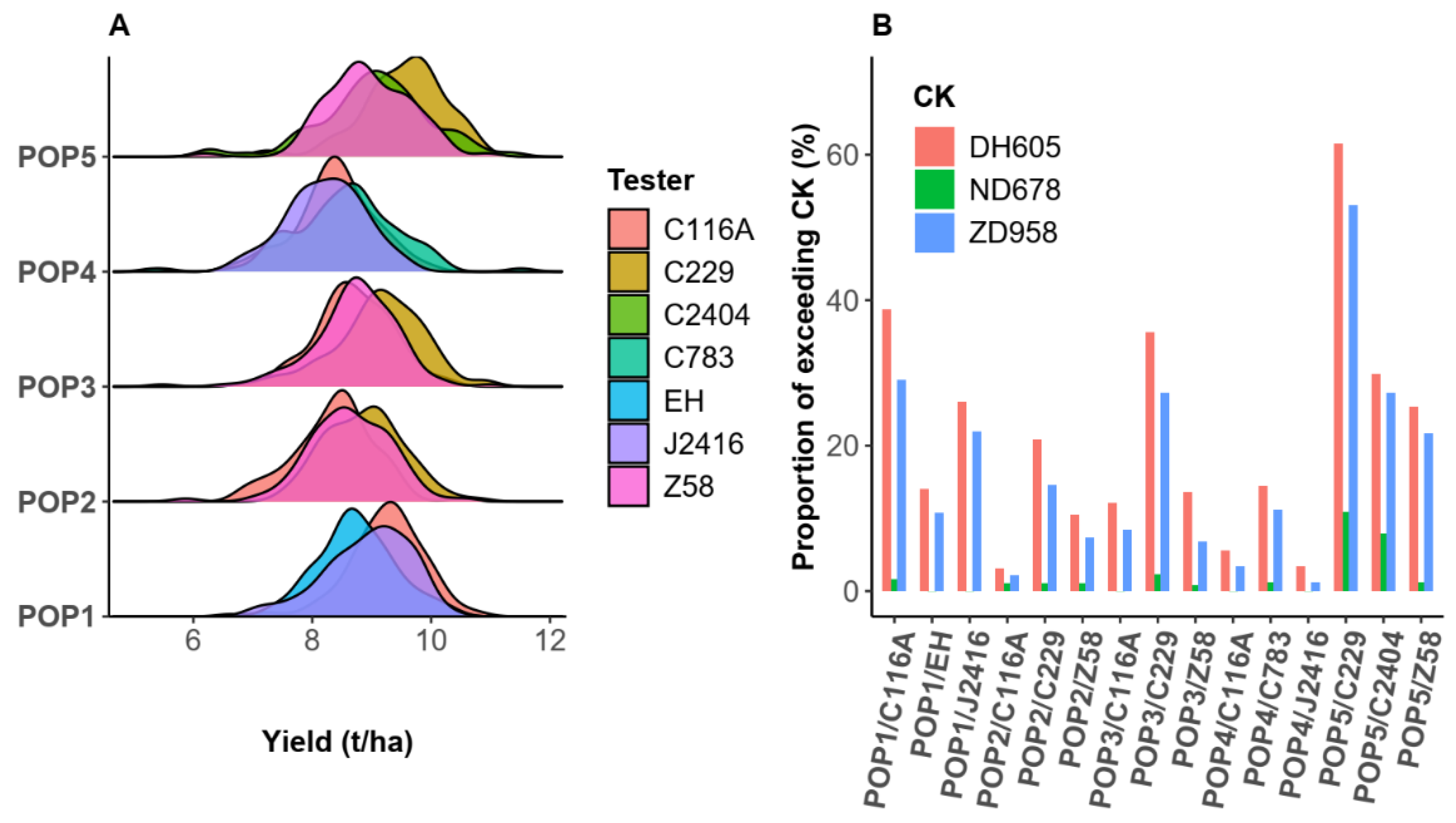
2.2. Basic Analysis of Phenotype and Effect of Cross Heterotic Group Fusion
2.3. Predicted Accuracy in DH and Hybrid Populations Using Different Models
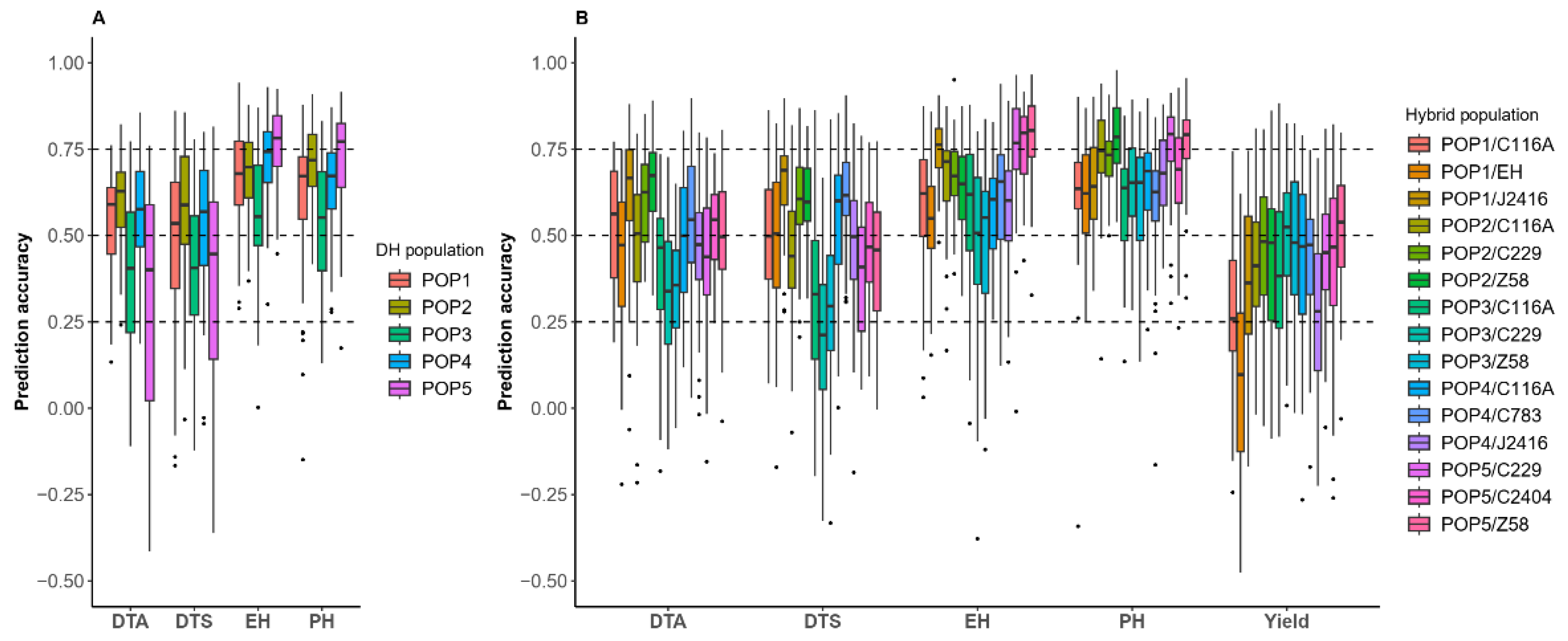
2.4. Prediction Accuracy of DH and Hybrid Within Populations for Different Traits
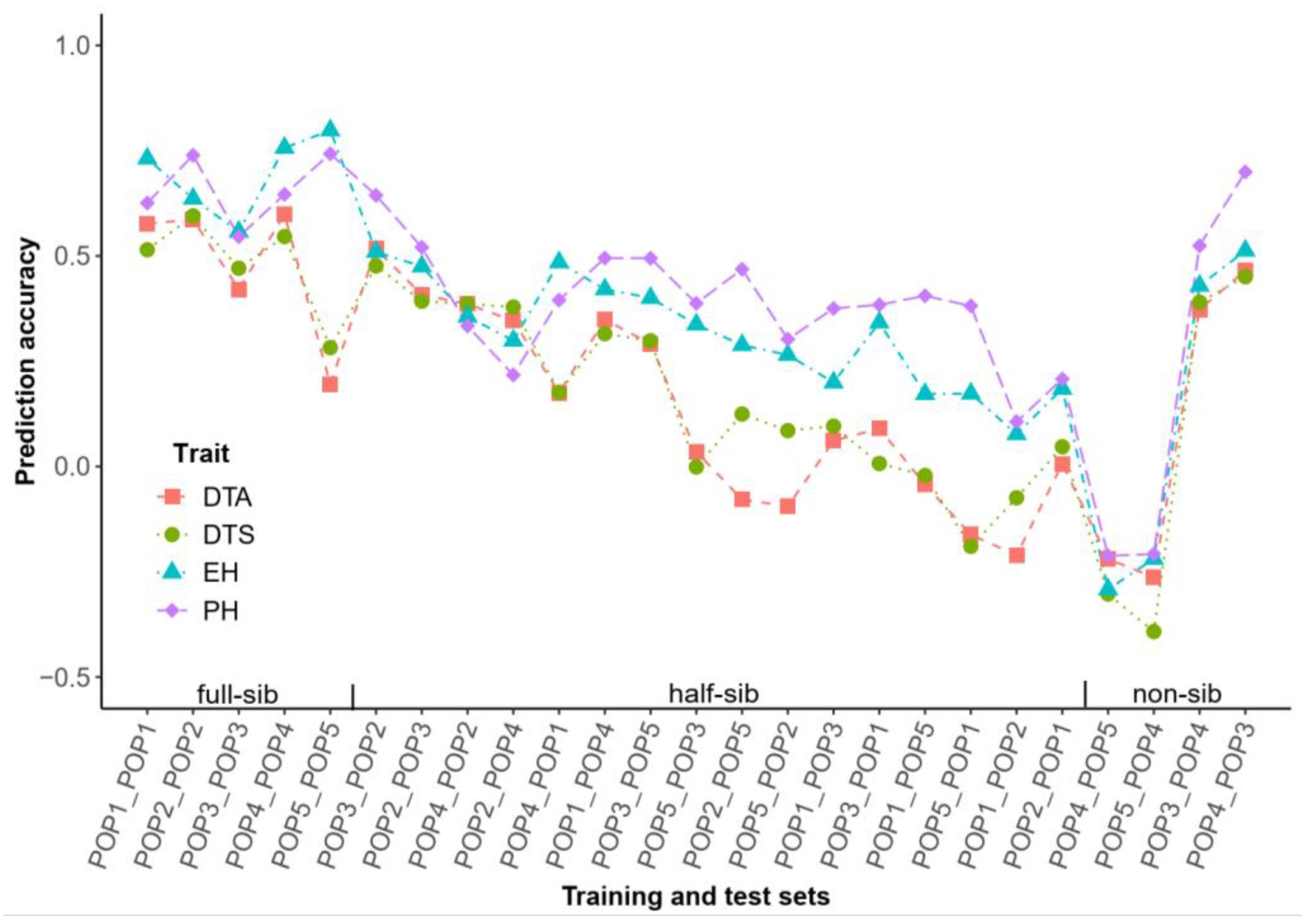
2.5. Prediction Accuracy of Cross DH Populations
2.6. Prediction Accuracy and Classification of Cross Hybrid Populations
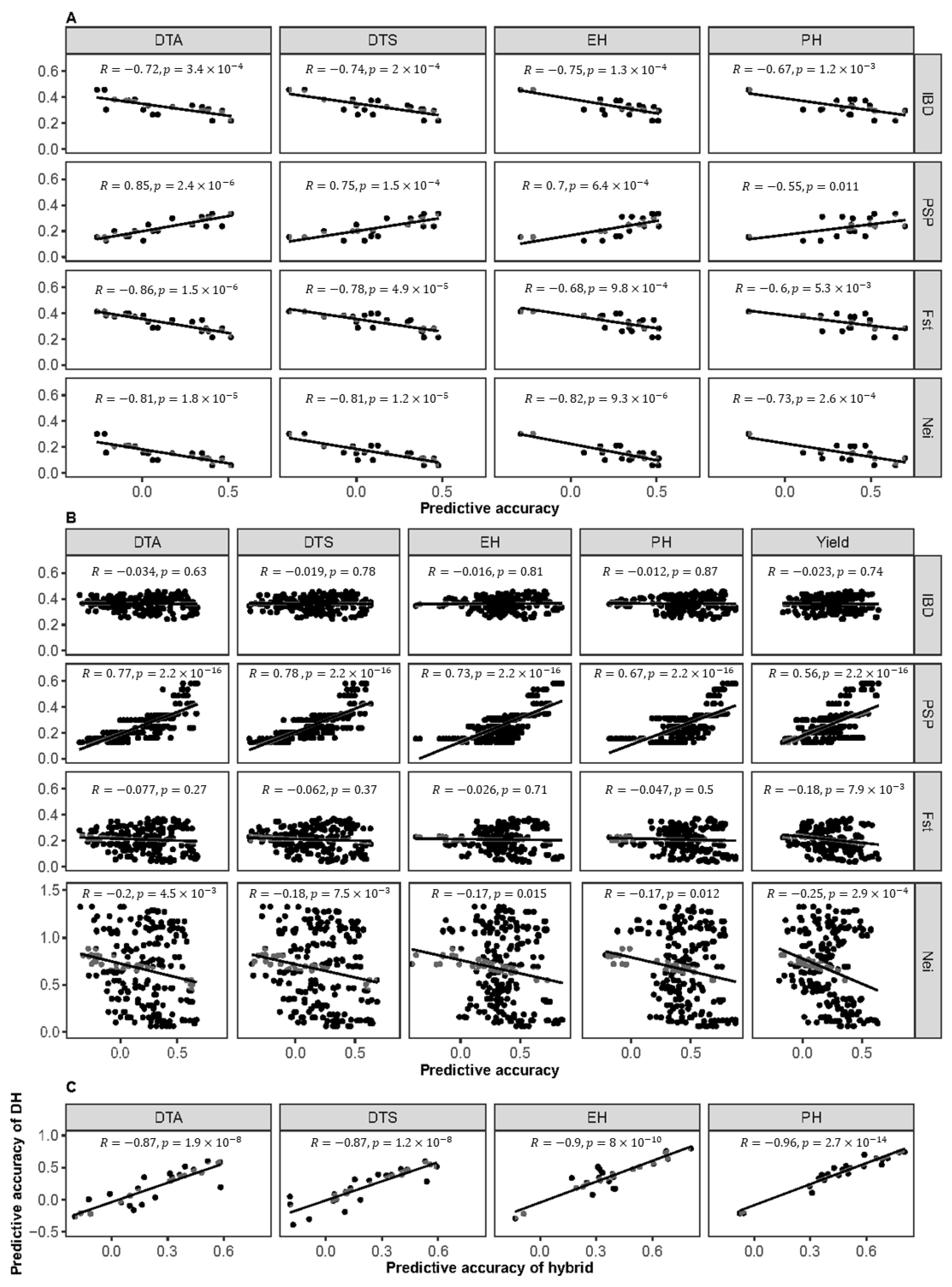
2.7. Relationship Between Prediction Accuracy and Genotypic Character of Cross DH or Cross Hybrid Populations
3. Discussion
3.1. Superior Hybrids Can Be Produced Through Inter-Heterotic-Group Crossing
3.2. RKHS Model Exploring the Best Prediction Accuracy Among the GS Models
3.3. Cross Population Prediction in Half-Sibs with Non-Shared Parents from the Same Heterotic Group Exhibiting Potential Accuracy in DH Population
3.4. Cross Population Prediction Among Hybrids from the Same DH Population Expressing Prominent Accuracy
3.5. The Proportion of Shared Polymorphisms (Considering SNPs) Between the Training and the Test Sets (PSP) Correlates with the Cross Population Prediction of Both the DH and Hybrid Populations
4. Materials and Methods
4.1. Plant Materials, Experimental Design, and Phenotypic Data Collection
4.2. Phenotypic Data Analysis and Heritability Estimation
4.3. Genotyping and Genotypic Data Analysis
4.4. Genomic Prediction
4.5. Calculation of Prediction Accuracy
4.6. DH Population Prediction Classification
4.7. Hybrid Population Prediction Type Division
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, J.; Wu, P.; Trampe, B.; Tian, X.; Lübberstedt, T.; Chen, S. Novel technologies in doubled haploid line development. Plant Biotechnol. J. 2017, 15, 1361–1370. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pérez-Rodríguez, P.; Semagn, K.; Beyene, Y.; Babu, R.; López-Cruz, M.A.; San Vicente, F.; Olsen, M.; Buckler, E.; Jannink, J.; et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity 2015, 114, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Beyene, Y.; Semagn, K.; Mugo, S.; Tarekegne, A.; Babu, R.; Meisel, B.; Sehabiague, P.; Makumbi, D.; Magorokosho, C.; Oikeh, S.; et al. Genetic Gains in Grain Yield Through Genomic Selection in Eight Bi-parental Maize Populations under Drought Stress. Crop Sci. 2015, 55, 154–163. [Google Scholar] [CrossRef]
- Crossa, J.; Perez, P.; Hickey, J.; Burgueno, J.; Ornella, L.; Ceron-Rojas, J.; Zhang, X.; Dreisigacker, S.; Babu, R.; Li, Y.; et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 2014, 112, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Charmet, G.; Storlie, E.; Oury, F.X.; Laurent, V.; Beghin, D.; Chevarin, L.; Lapierre, A.; Perretant, M.R.; Rolland, B.; Heumez, E.; et al. Genome-wide prediction of three important traits in bread wheat. Mol. Breed. 2014, 34, 1843–1852. [Google Scholar] [CrossRef]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef]
- Xu, S.; Zhu, D.; Zhang, Q. Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc. Natl. Acad. Sci. USA 2014, 111, 12456–12461. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Yang, Z.; Zheng, X.; Yu, S.; Xu, C.; Hu, Z. Predicting rice hybrid performance using univariate and multivariate GBLUP models based on North Carolina mating design II. Heredity 2017, 118, 302–310. [Google Scholar] [CrossRef]
- Sallam, A.H.; Endelman, J.B.; Jannink, J.L.; Smith, K.P. Assessing Genomic Selection Prediction Accuracy in a Dynamic Barley Breeding Population. Plant Genome 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Schmidt, M.; Kollers, S.; Maasberg-Prelle, A.; Großer, J.; Schinkel, B.; Tomerius, A.; Graner, A.; Korzun, V. Prediction of malting quality traits in barley based on genome-wide marker data to assess the potential of genomic selection. Theor. Appl. Genet. 2016, 129, 203–213. [Google Scholar] [CrossRef]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef]
- Wong, C.K.; Bernardo, R. Genomewide selection in oil palm: Increasing selection gain per unit time and cost with small populations. Theor. Appl. Genet. 2008, 116, 815–824. [Google Scholar] [CrossRef]
- Resende, M.F.R.; Muñoz, P.; Resende, M.D.V.; Garrick, D.J.; Fernando, R.L.; Davis, J.M.; Jokela, E.J.; Martin, T.A.; Peter, G.F.; Kirst, M. Accuracy of Genomic Selection Methods in a Standard Data Set of Loblolly Pine (Pinus taeda L.). Genetics 2012, 190, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Isik, F.; Bartholomé, J.; Farjat, A.; Chancerel, E.; Raffin, A.; Sanchez, L.; Plomion, C.; Bouffier, L. Genomic selection in maritime pine. Plant Sci. 2016, 242, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Combs, E.; Bernardo, R. Accuracy of Genomewide Selection for Different Traits with Constant Population Size, Heritability, and Number of Markers. Plant Genome 2013, 6. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.L.; Iwata, H.; Souza, E.; Sorrells, M.E. Genomic Selection Accuracy for Grain Quality Traits in Biparental Wheat Populations. Crop Sci. 2011, 51, 2597–2606. [Google Scholar] [CrossRef]
- Elsen, J.M. Approximated prediction of genomic selection accuracy when reference and candidate populations are related. Genet. Sel. Evol. 2016, 48, 18. [Google Scholar] [CrossRef]
- Pszczola, M.; Strabel, T.; Mulder, H.A.; Calus, M.P.L. Reliability of direct genomic values for animals with different relationships within and to the reference population. J. Dairy Sci. 2012, 95, 389–400. [Google Scholar] [CrossRef]
- Edwards, S.M.; Buntjer, J.B.; Jackson, R.; Bentley, A.R.; Lage, J.; Byrne, E.; Burt, C.; Jack, P.; Berry, S.; Flatman, E.; et al. The effects of training population design on genomic prediction accuracy in wheat. Theor. Appl. Genet. 2019, 132, 1943–1952. [Google Scholar] [CrossRef]
- 21. Whittaker, J.C.; Thompson, R.; Denham, M.C. Marker-assisted selection using ridge regression. Genet. Res. 2000, 75, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Zhang, X.; Pérez-Rodríguez, P.; Burgueño, J.; Olsen, M.; Buckler, E.; Atlin, G.; Prasanna, B.M.; Vargas, M.; San Vicente, F.; Crossa, J. Rapid Cycling Genomic Selection in a Multiparental Tropical Maize Population. G3 Genes Genomes Genetics. 2017, 7, 2315–2326. [Google Scholar] [CrossRef]
- Juliana, P.; Singh, R.P.; Singh, P.K.; Crossa, J.; Rutkoski, J.E.; Poland, J.A.; Bergstrom, G.C.; Sorrells, M.E. Comparison of Models and Whole-Genome Profiling Approaches for Genomic-Enabled Prediction of Septoria Tritici Blotch, Stagonospora Nodorum Blotch, and Tan Spot Resistance in Wheat. Plant Genome 2017, 10. [Google Scholar] [CrossRef]
- Bernardo, R. Best Linear Unbiased Prediction of Maize Single-Cross Performance. Crop Sci. 1996, 36, 50–56. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Heslot, N.; Yang, H.P.; Sorrells, M.E.; Jannink, J.L. Genomic Selection in Plant Breeding: A Comparison of Models. Crop Sci. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- de Los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Gianola, D. Priors in whole-genome regression: The bayesian alphabet returns. Genetics 2013, 194, 573–596. [Google Scholar] [CrossRef]
- de Los, C.G.; Gianola, D.; Rosa, G.J. Reproducing kernel Hilbert spaces regression: A general framework for genetic evaluation. J. Anim. Sci. 2009, 87, 1883–1887. [Google Scholar] [CrossRef]
- De Los Campos, G.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A.; Crossa, J. Semi-parametric genomic-enabled prediction of genetic values using reproducing kernel Hilbert spaces methods. Genet. Res. 2010, 92, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Camacho, J.M.; de Los, C.G.; Perez, P.; Gianola, D.; Cairns, J.E.; Mahuku, G.; Babu, R.; Crossa, J. Genome-enabled prediction of genetic values using radial basis function neural networks. Theor. Appl. Genet. 2012, 125, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Camacho, J.M.; Crossa, J.; Perez-Rodriguez, P.; Ornella, L.; Gianola, D. Genome-enabled prediction using probabilistic neural network classifiers. BMC Genom. 2016, 17, 208. [Google Scholar] [CrossRef]
- Ornella, L.; Pérez, P.; Tapia, E.; González-Camacho, J.M.; Burgueño, J.; Zhang, X.; Singh, S.; Vicente, F.S.; Bonnett, D.; Dreisigacker, S.; et al. Genomic-enabled prediction with classification algorithms. Heredity 2014, 112, 616–626. [Google Scholar] [CrossRef]
- Zhao, Y.; Gowda, M.; Liu, W.; Würschum, T.; Maurer, H.P.; Longin, F.H.; Ranc, N.; Reif, J.C. Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet. 2012, 124, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tucker, D.M.; Basten, C.J.; Gandhi, H.; Ersoz, E.; Guo, B.; Xu, Z.; Wang, D.; Gay, G. The impact of population structure on genomic prediction in stratified populations. Theor. Appl. Genet. 2014, 127, 749–762. [Google Scholar] [CrossRef]
- Isidro, J.; Jannink, J.L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.; McCouch, S.R. Genomic Selection and Association Mapping in Rice (Oryza sativa): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef]
- Tracy, W.F.; Chandler, M.A. The Historical and Biological Basis of the Concept of Heterotic Patterns in Corn Belt Dent Maize. In Plant Breeding: The Arnel R. Hallauer International Symposium; Blackwell Publishing: Ames, IA, USA, 2008. [Google Scholar]
- Zhao, J.; Li, C.; Song, W.; Wang, Y.; Xing, J.; Zhang, R.; Yi, H.; Yang, Y.; Shi, Z.; Wang, J. Elaboration of Heterotic Pattern in a Series of Maize Varieties by SSR Markers. J. Maize Sci. 2017, 25, 1–8. [Google Scholar] [CrossRef]
- Luo, J.; Wang, M.; Jia, G.; He, Y. Transcriptome-wide analysis of epitranscriptome and translational efficiency associated with heterosis in maize. J. Exp. Bot. 2021, 72, 2933–2946. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Breeding Potential of Intra- and Interheterotic Group Crosses in Maize. Crop Sci. 2001, 41, 68–71. [Google Scholar] [CrossRef]
- Pan, G.; Yang, K.; Li, W.; Huang, Y.; Gao, S.; Lan, H.; Li, L.; Cao, M.; Tang, Q.; Fu, F.; et al. A Review of the Research and Application of Heterotic Groups and Patterns of Maize Breeding in Southwest China. J. Maize Sci. 2020, 28, 1–8. [Google Scholar]
- Carena, M.J.; Hallauer, A.R.; Miranda Filho, J.B. Quantitative Genetics in Maize Breeding; Springer: New York, NY, USA, 2010. [Google Scholar]
- Xu, Y.; Wang, X.; Ding, X.; Zheng, X.; Yang, Z.; Xu, C.; Hu, Z. Genomic selection of agronomic traits in hybrid rice using an NCII population. Rice 2018, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Azodi, C.B.; Bolger, E.; McCarren, A.; Roantree, M.; de Los Campos, G.; Shiu, S. Benchmarking Parametric and Machine Learning Models for Genomic Prediction of Complex Traits. G3 Genes Genomes Genetics. 2019, 9, 3691–3702. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Villanueva, B.; Woolliams, J.A.; Weedon, M.N. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 2008, 3, e3395. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic Selection Accuracy using Multifamily Prediction Models in a Wheat Breeding Program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Albrecht, T.; Wimmer, V.; Auinger, H.; Erbe, M.; Knaak, C.; Ouzunova, M.; Simianer, H.; Schön, C. Genome-based prediction of testcross values in maize. Theor. Appl. Genet. 2011, 123, 339–350. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Endelman, J.B.; Stange, M.; Sorrells, M.E.; Jannink, J.L.; Melchinger, A.E. Genomic predictability of interconnected biparental maize populations. Genetics 2013, 194, 493–503. [Google Scholar] [CrossRef]
- Schopp, P.; Müller, D.; Wientjes, Y.C.J.; Melchinger, A.E. Genomic Prediction Within and Across Biparental Families: Means and Variances of Prediction Accuracy and Usefulness of Deterministic Equations. G3 Genes Genomes Genet. 2017, 7, 3571–3586. [Google Scholar] [CrossRef]
- van den Berg, S.; Calus, M.P.; Meuwissen, T.H.; Wientjes, Y.C. Across population genomic prediction scenarios in which Bayesian variable selection outperforms GBLUP. BMC Genet. 2015, 16, 146. [Google Scholar] [CrossRef]
- Wientjes, Y.; Veerkamp, R.F.; Bijma, P.; Bovenhuis, H.; Schrooten, C.; Calus, M. Empirical and deterministic accuracies of across-population genomic prediction. Genet. Sel. Evol. 2015, 47, 5. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The Impact of Genetic Relationship Information on Genome-Assisted Breeding Values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Arguello-Blanco, M.N.; Sneller, C.H. The effect of cycles of genomic selection on the wheat (T. aestivum) genome. Theor. Appl. Genet. 2023, 136, 70. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Maenhout, S.; De Baets, B.; Haesaert, G.; Van Bockstaele, E. Support vector machine regression for the prediction of maize hybrid performance. Theor. Appl. Genet. 2007, 115, 1003–1013. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Statistics, L.B.; Breiman, L. Random Forests. Mach. Learn. 2001, 5–32. [Google Scholar]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Gianola, D.; Okut, H.; Weigel, K.A.; Rosa, G.J. Predicting complex quantitative traits with Bayesian neural networks: A case study with Jersey cows and wheat. BMC Genet. 2011, 12, 87. [Google Scholar] [CrossRef]
- Choudhary, R.; Gianey, H.K. Comprehensive Review On Supervised Machine Learning Algorithms. In Proceedings of the 2017 International Conference on Machine Learning and Data Science (MLDS), Noida, India, 14–15 December 2017; pp. 37–43. [Google Scholar]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Meyer, D.; Dimitriadou, E.; Hornik, K.; Weingessel, A.; Leisch, F. Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien [R package e1071 version 1.7-16]. 2020. Available online: https://CRAN.R-project.org/package=e1071 (accessed on 1 October 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prediction Scenario | Trait | RKHS | BayesB | RRBLUP | SVM | RF |
|---|---|---|---|---|---|---|
| Prediction within DH populations | DTA | 0.52 | 0.54 | 0.51 | 0.49 | 0.44 |
| DTS | 0.51 | 0.49 | 0.47 | 0.49 | 0.44 | |
| EH | 0.69 | 0.69 | 0.69 | 0.59 | 0.56 | |
| PH | 0.68 | 0.68 | 0.67 | 0.57 | 0.56 | |
| Average | 0.60 | 0.60 | 0.58 | 0.53 | 0.50 | |
| Prediction within hybrid populations | DTA | 0.51 | 0.51 | 0.50 | 0.47 | 0.48 |
| DTS | 0.48 | 0.48 | 0.48 | 0.45 | 0.47 | |
| EH | 0.66 | 0.66 | 0.65 | 0.56 | 0.56 | |
| PH | 0.69 | 0.69 | 0.70 | 0.56 | 0.58 | |
| Yield | 0.41 | 0.41 | 0.39 | 0.35 | 0.36 | |
| Average | 0.56 | 0.56 | 0.56 | 0.48 | 0.49 | |
| Cross population prediction of DH | DTA | 0.19 | 0.19 | 0.19 | 0.21 | 0.18 |
| DTS | 0.20 | 0.20 | 0.20 | 0.21 | 0.20 | |
| EH | 0.36 | 0.36 | 0.36 | 0.29 | 0.28 | |
| PH | 0.41 | 0.39 | 0.41 | 0.35 | 0.33 | |
| Average | 0.29 | 0.28 | 0.29 | 0.27 | 0.25 | |
| Cross population prediction of hybrid | DTA | 0.23 | 0.23 | 0.22 | 0.21 | 0.22 |
| DTS | 0.21 | 0.22 | 0.20 | 0.20 | 0.21 | |
| EH | 0.37 | 0.37 | 0.36 | 0.31 | 0.32 | |
| PH | 0.45 | 0.43 | 0.45 | 0.38 | 0.39 | |
| Yield | 0.23 | 0.24 | 0.21 | 0.22 | 0.22 | |
| Average | 0.32 | 0.31 | 0.31 | 0.28 | 0.28 | |
| Overall mean | 0.44 | 0.44 | 0.43 | 0.39 | 0.38 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, D.; Li, J.; Guo, S.; Wang, Y.; Xu, S.; Chen, S.; Liu, W. Genomic Prediction for Germplasm Improvement Through Inter-Heterotic-Group Line Crossing in Maize. Int. J. Mol. Sci. 2025, 26, 2662. https://doi.org/10.3390/ijms26062662
Cheng D, Li J, Guo S, Wang Y, Xu S, Chen S, Liu W. Genomic Prediction for Germplasm Improvement Through Inter-Heterotic-Group Line Crossing in Maize. International Journal of Molecular Sciences. 2025; 26(6):2662. https://doi.org/10.3390/ijms26062662
Chicago/Turabian StyleCheng, Dehe, Jinlong Li, Shuwei Guo, Yuandong Wang, Shizhong Xu, Shaojiang Chen, and Wenxin Liu. 2025. "Genomic Prediction for Germplasm Improvement Through Inter-Heterotic-Group Line Crossing in Maize" International Journal of Molecular Sciences 26, no. 6: 2662. https://doi.org/10.3390/ijms26062662
APA StyleCheng, D., Li, J., Guo, S., Wang, Y., Xu, S., Chen, S., & Liu, W. (2025). Genomic Prediction for Germplasm Improvement Through Inter-Heterotic-Group Line Crossing in Maize. International Journal of Molecular Sciences, 26(6), 2662. https://doi.org/10.3390/ijms26062662