
Precision Medicine in High-Grade Serous Ovarian Cancer: Targeted Therapies and the Challenge of Chemoresistance

Abstract
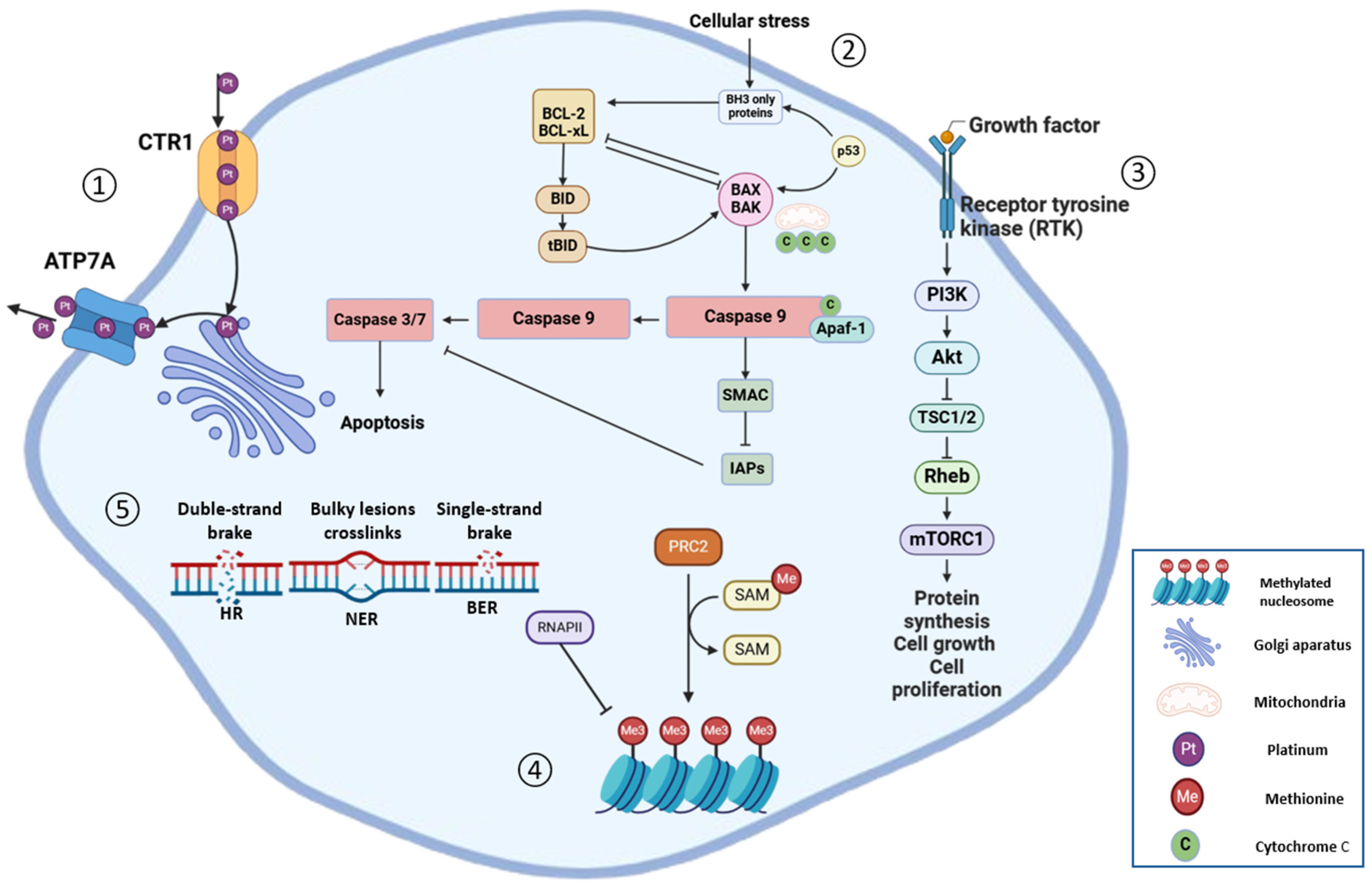
:1. Introduction
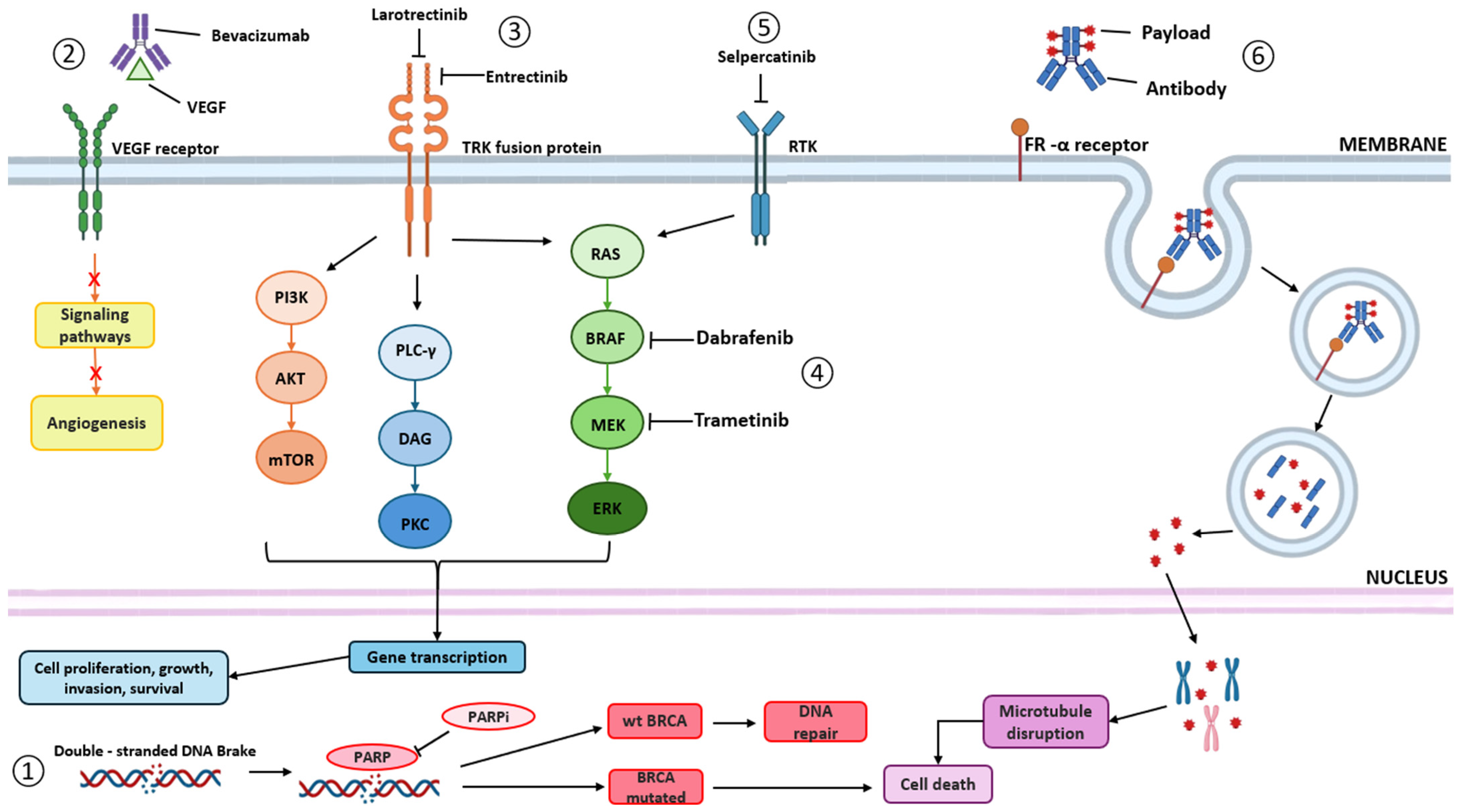
2. Approved Targeted Therapies—Pros and Cons
2.1. First-Line and Maintenance Therapies
2.1.1. Poly (Adenosine Diphosphate-Ribose) Polymerase Inhibitors (PARPis)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | FDA Approved | EMA Approved | References |
|---|---|---|---|
| Olaparib | ✓ | ✓ | [20,34] |
| Niraparib | ✓ | ✓ | [22,35] |
| Rucaparib | ✓ | ✓ | [24,36] |
| Bevacizumab | ✓ | ✓ | [18,55] |
| Larotrectinib | ✓ | ✓ | [25,26] |
| Entrectinib | ✓ | ✓ | [28,56] |
| Dabrafenib + Trametinib | ✓ | ✕ | [31] |
| Selpercatinib | ✓ | ✓ | [57,58] |
| Mirvetuximab Soravtansine | ✓ | ✓ | [32,33] |
2.1.2. Bevacizumab
2.2. Therapies for Persistent and Recurrent Disease
2.2.1. TRK Inhibitors
Larotrectinib
Entrectinib
2.2.2. RET Inhibitors
Selpercatinib
2.2.3. BRAF Inhibitors
2.2.4. Antibody–Drug Conjugates (ADCs)
Mirvetuximab Soravtansine
3. Promising Innovative Therapies
3.1. ICIs in Combination with PARPis and Anti-Angiogenic Agents
3.2. PI3K Inhibitors
3.3. WEE1 Inhibitors
3.4. ATR Inhibitors
| Targeted Therapy | Mechanism of Action | Target | Potential Benefits | Clinical Trials | References |
|---|---|---|---|---|---|
| Immune Checkpoint Inhibitors | Block proteins that act as brakes on the immune system | CTLA-4, PD-1, and PD-L1 | Enhance the ability of T cells to recognize and attack cancer cells | OPEB-01/APGOT-OV4 (NCT04361370) MEDIOLA (NCT02734004) ATHENA-COMBO (NCT03522246) | [69,124,129] |
| Anti-angiogenic Agents | Inhibit the growth of new blood vessels (angiogenesis) that supply tumours | VEGF (vascular endothelial growth factor) and its receptors | Starve the tumour of nutrients and oxygen, inhibiting its growth | CONCERTO (NCT02889900) | [5,118,147] |
| PI3K Inhibitors | Block the phosphoinositide 3-kinase (PI3K) pathway | PI3K enzymes involved in cell growth, proliferation, and survival | Reduce cancer cell proliferation and induce apoptosis | EPIK-O/ENGOT-OV16 (NCT04729387) | [132,133] |
| WEE1 Inhibitors | Inhibit WEE1 kinase, a regulator of the cell cycle | WEE1, which controls the G2/M checkpoint | Force cancer cells to enter mitosis prematurely, leading to cell death due to DNA damage | Adavosertib monotherapy (NCT02482311) Adavosertib + carboplatin (NCT01164995) | [136,137,139] |
| ATR Inhibitors | Inhibit ATR (ataxia telangiectasia and Rad3-related protein) kinase | ATR, involved in the DNA damage response | Prevent cancer cells from repairing DNA damage, leading to cell death | CAPRI (NTC03462342) | [144,145] |
4. Predictive Biomarkers for Platinum-Resistant Tumours
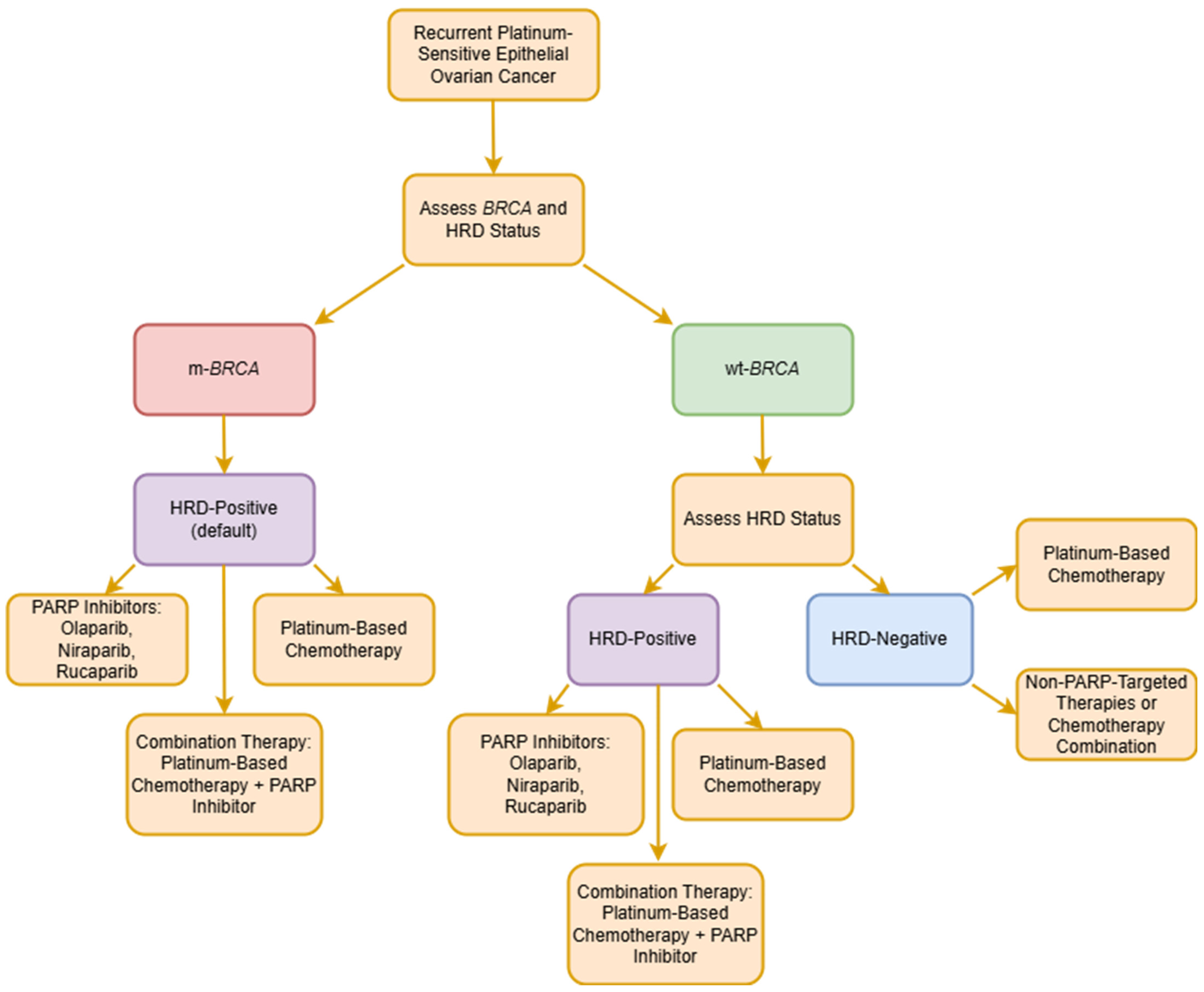
4.1. HRD Status
4.2. Epigenetic Modification
4.3. MicroRNAs
4.4. TME Factors
4.5. TP53 Mutations
5. Successes and Challenges in Precision Medicine
| Treatment | Estimated Cost per Test (US) | Estimated Cost per Test (Europe) | Notes | References |
|---|---|---|---|---|
| BRCA Testing | USD 1500–3000 | EUR 1100–2500 | Costs can vary based on the laboratory and specific testing methods used. | [187,188] |
| Homologous Recombination Deficiency (HRD) Testing | USD 2500–4000 | EUR 1800–3000 | HRD testing may include BRCA testing as part of a broader panel. | [188,189] |
| Microsatellite Instability (MSI) Testing | USD 500–2000 | EUR 300–1500 | MSI testing can be performed via PCR or NGS methods, influencing cost. | [190] |
| Chromosomal Instability (CIN) Testing | USD 2000–3000 | EUR 1500–2500 | CIN testing methods and availability may vary, affecting cost. | [191] |
| Whole-Genome Sequencing (WGS) | USD 5000–10,000 | EUR 4000–8000 | WGS offers extensive data but at a higher cost and longer turnaround time. | [192] |
| Whole-Exome Sequencing (WES) | USD 1500–5000 | EUR 400–4000 | WES is less comprehensive than WGS but more cost-effective and faster. | [192] |
| Tumour Mutational Burden (TMB) Testing | USD 1500–3000 | EUR 1–2500 | TMB can be assessed via targeted gene panels or broader sequencing approaches. | [193] |
| Surgery | USD 95,000 | EUR 35,000 | Surgical costs depend on the procedure’s complexity, hospital fees, and geographic location. In the EU, prices vary by country and healthcare system. | [194,195] |
| Carboplatin | USD 125–500 | EUR 100–250 | Carboplatin is a generic chemotherapy drug, making it relatively affordable. Prices depend on dosage and healthcare provider. | [189] |
| Rucaparib (Rubraca) | USD 10,000–16,000 | EUR 8000–11,500 | A PARPi used for maintenance therapy in recurrent ovarian cancer. Costs are based on average monthly expenses. | [196] |
| Olaparib (Lynparza) | USD 10,000–16,000 | EUR 5200–13,000 | Another PARPi approved for BRCA-mutated ovarian cancer. Monthly costs are similar to rucaparib. | [188,196] |
| Niraparib (Zejula) | USD 10,000–18,000 | EUR 5500–14,800 | A PARPi approved for maintenance treatment in ovarian cancer. Monthly costs are comparable to other PARPis. | [188,196] |
| Bevacizumab (Avastin) | USD 3000–13,000 | EUR 4200–8400 | An angiogenesis inhibitor used in combination with chemotherapy for ovarian cancer. Costs vary based on dosage and treatment duration. | [189] |
| Larotrectinib (Vitrakvi) | USD 32,800 | EUR 28,000 | Approved for tumours with NTRK gene fusions, including some ovarian cancers. | [197] |
| Entrectinib (Rozlytrek) | USD 17,050 | EUR 14,500 | Similar to larotrectinib, targets NTRK gene fusions. | [197] |
| Dabrafenib (Tafinlar) | USD 12,000–15,000 | EUR 6000–7000 | Targets BRAF-mutated cancers; used in combination with trametinib for certain ovarian cancers. | [198] |
| Trametinib (Mekinist) | USD 12,000–15,000 | EUR 6000–7000 | MEK inhibitor often used with dabrafenib. Monthly cost is similar to dabrafenib. | [198] |
| Selpercatinib (Retevmo) | USD 20,600 | EUR 17,600 | RET inhibitor approved for specific gene mutations; its use in ovarian cancer is under investigation. | [199] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dalmartello, M.; La Vecchia, C.; Bertuccio, P.; Boffetta, P.; Levi, F.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2022 with focus on ovarian cancer. Ann. Oncol. 2022, 33, 330–339. [Google Scholar] [CrossRef]
- GLOBOCAN. Global Cancer Observatory. Cancer Today. 2025. Available online: https://gco.iarc.fr/today/en/dataviz/tables?mode=population&cancers=25&types=1 (accessed on 19 February 2025).
- Lukanović, D.; Herzog, M.; Kobal, B.; Černe, K. The contribution of copper efflux transporters ATP7A and ATP7B to chemoresistance and personalized medicine in ovarian cancer. Biomed. Pharmacother. 2020, 129, 110401. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Yang, L.; Xie, H.J.; Li, Y.Y.; Wang, X.; Liu, X.X. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar]
- Colombo, N.; Tomao, F.; Benedetti Panici, P.; Nicoletto, M.O.; Tognon, G.; Bologna, A.; Lissoni, A.A.; DeCensi, A.; Lapresa, M.; Mancari, R.; et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol. Oncol. 2022, 164, 505–513. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Women’s Health 2019, 11, 287–299. [Google Scholar] [CrossRef]
- Huang, J.; Chan, W.C.; Ngai, C.H.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E.; Xu, W.; Zheng, Z.J.; Elcarte, E.; Withers, M.; et al. Worldwide Burden, Risk Factors, and Temporal Trends of Ovarian Cancer: A Global Study. Cancers 2022, 14, 2230. [Google Scholar] [CrossRef]
- Rosenberg, S.M.; Ruddy, K.J.; Tamimi, R.M.; Gelber, S.; Schapira, L.; Come, S.; Borges, V.F.; Larsen, B.; Garber, J.E.; Partridge, A.H. BRCA1 and BRCA2 mutation testing in young women with breast cancer. JAMA Oncol. 2016, 2, 730–736. [Google Scholar] [CrossRef]
- Sledge, G.W. What is Targeted Therapy? J. Clin. Oncol. 2005, 23, 1614–1615. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Lukanović, D.; Kobal, B.; Černe, K. Ovarian Cancer: Treatment and Resistance to Pharmacotherapy. Reprod. Med. 2022, 3, 127–140. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.M.; Ray-Coquard, I.; Moore, K.N.; Diéras, V.; Alexandre, J. PARP inhibitors and newly second primary malignancies in cancer patients: A systematic review and safety meta-analysis of placebo randomized controlled trials. Ann. Oncol. 2021, 32, 1048–1050. [Google Scholar] [CrossRef]
- Brunton, L.L.; Hilal-Dandan, R.; Knollmann, B.C. (Eds.) Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th ed.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Ledermann, J.A.; Matias-Guiu, X.; Amant, F.; Concin, N.; Davidson, B.; Fotopoulou, C.; González-Martin, A.; Gourley, C.; Leary, A.; Lorusso, D.; et al. ESGO–ESMO–ESP consensus conference recommendations on ovarian cancer: Pathology and molecular biology and early, advanced and recurrent disease. Ann. Oncol. 2024, 35, 248–266. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Guidlines. Ovarian Cancer/Fallopian Tube Cancer/Primary Peritoneal Cancer (Version 1.2024). 2024. Available online: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed on 8 May 2024).
- EMA. European Medicines Agency. Avastin: EPAR—Product Information. 2023. Available online: https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Olaparib Plus Bevacizumab as Maintenance Treatment for Ovarian, Fallopian Tube, or Primary Peritoneal Cancers. 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary (accessed on 5 September 2024).
- EMA. European Medicines Agency. Lynparza: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/lynparza-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Olaparib Tablets for Maintenance Treatment in Ovarian Cancer. 2017. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-tablets-maintenance-treatment-ovarian-cancer (accessed on 5 September 2024).
- EMA. European Medicines Agency. Zejula: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/zejula-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Maintenance Treatment for Recurrent Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancers. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-maintenance-treatment-recurrent-epithelial-ovarian-fallopian-tube-or-primary-peritoneal (accessed on 5 September 2024).
- EMA. European Medicines Agency. Rubraca: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/rubraca-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Rucaparib for Maintenance Treatment of Recurrent Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-rucaparib-maintenance-treatment-recurrent-ovarian-fallopian-tube-or-primary-peritoneal (accessed on 5 September 2024).
- EMA. European Medicines Agency. Vitrakvi: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/vitrakvi-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. 2018. Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions-0 (accessed on 25 April 2024).
- EMA. European Medicines Agency. Rozlytrek: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/rozlytrek-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC. 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 25 April 2024).
- EMA. European Medicines Agency. Retsevmo: EPAR—Product Information. 2025. Available online: https://www.ema.europa.eu/en/documents/product-information/retsevmo-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Grants Accelerated Approval to Dostarlimab-gxly for dMMR Advanced Solid Tumors. 2023. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors (accessed on 30 October 2024).
- FDA. Food and Drug Administration. FDA Grants Accelerated Approval to Dabrafenib in Combination with Trametinib for Unresectable or Metastatic Solid Tumors with BRAF V600E Mutation. 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid (accessed on 8 May 2024).
- EMA. European Medicines Agency. Elahere: EPAR—Product Information. 2024. Available online: https://www.ema.europa.eu/en/documents/product-information/elahere-epar-product-information_en.pdf (accessed on 18 February 2025).
- FDA. Food and Drug Administration. FDA Approves Mirvetuximab Soravtansine-gynx for FRα Positive, Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. 2024. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-mirvetuximab-soravtansine-gynx-fra-positive-platinum-resistant-epithelial-ovarian (accessed on 21 February 2025).
- EMA. Europena Medicines Agency. Lynparza. 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza (accessed on 24 June 2024).
- EMA. European Medicines Agency. Zejula. 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zejula (accessed on 24 June 2024).
- EMA. European Medicines Agency. Rubraca. 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rubraca (accessed on 24 June 2024).
- Drugs.com. Lynparza FDA Approval History. 2023. Available online: https://www.drugs.com/history/lynparza.html (accessed on 24 June 2024).
- Drugs.com. Zejula FDA Approval History. 2023. Available online: https://www.drugs.com/history/zejula.html (accessed on 24 June 2024).
- Drugs.com. Rubraca FDA Approval History. 2021. Available online: https://www.drugs.com/history/rubraca.html (accessed on 24 June 2024).
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Morales, J.C.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef]
- Bound, N.T.; Vandenberg, C.J.; Kartikasari, A.E.R.; Plebanski, M.; Scott, C.L. Improving PARP inhibitor efficacy in high-grade serous ovarian carcinoma: A focus on the immune system. Front. Genet. 2022, 13, 886170. [Google Scholar] [CrossRef] [PubMed]
- Cordani, N.; Bianchi, T.; Ammoni, L.C.; Cortinovis, D.L.; Cazzaniga, M.E.; Lissoni, A.A.; Landoni, F.; Canova, S. An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective. Int. J. Mol. Sci. 2023, 24, 11890. [Google Scholar] [CrossRef] [PubMed]
- Tuninetti, V.; Marín-Jiménez, J.A.; Valabrega, G.; Ghisoni, E. Long-term outcomes of PARP inhibitors in ovarian cancer: Survival, adverse events, and post-progression insights. ESMO Open 2024, 9, 103984. [Google Scholar] [CrossRef]
- Incorvaia, L.; Perez, A.; Marchetti, C.; Brando, C.; Gristina, V.; Cancelliere, D.; Pivetti, A.; Contino, S.; Di Giovanni, E.; Barraco, N.; et al. Theranostic biomarkers and PARP-inhibitors effectiveness in patients with non-BRCA associated homologous recombination deficient tumors: Still looking through a dirty glass window? Cancer Treat. Rev. 2023, 21, 102650. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ou, S.; Wei, H.; Qin, X.; Jiang, Q. Comparative Efficacy and Safety of Poly (ADP-Ribose) Polymerase Inhibitors in Patients with Ovarian Cancer: A Systematic Review and Network Meta-Analysis. Front. Oncol. 2022, 12, 815265. [Google Scholar] [CrossRef]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in parp inhibitors for the treatment of ovarian cancer: Mechanisms of action, pharmacology, safety, and efficacy. Int. J. Mol. Sci. 2021, 22, 4203. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. Phosphatase 1 nuclear targeting subunit, a novel DNA repair partner of PARP1. Cancer Res. 2019, 79, 2460–2461. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, B.; Lee, J.M.; Lheureux, S.; Liu, F.; et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Heo, Y.A.; Dhillon, S. Olaparib Tablet: A Review in Ovarian Cancer Maintenance Therapy. Target. Oncol. 2018, 13, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Parkinson, C.; Cheol Lim, M.; O’Malley, D.M.; Oaknin, A.; Wilson, M.K.; Coleman, R.L.; Lorusso, D.; Bessette, P.; Ghamande, S.; et al. A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients with Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 2022, 40, 3952–3964. [Google Scholar] [CrossRef]
- Friedlander, M.; Lee, Y.C.; Tew, W.P. Managing Adverse Effects Associated with Poly (ADP-ribose) Polymerase Inhibitors in Ovarian Cancer: A Synthesis of Clinical Trial and Real-World Data. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390876. [Google Scholar] [CrossRef]
- EMA. European Medicines Agency. Avastin. 2017. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/avastin (accessed on 5 September 2024).
- EMA. European Medicines Agency. Rozlytrek. 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/rozlytrek (accessed on 5 September 2024).
- FDA. Food and Drug Administration. FDA Approves Selpercatinib for Locally Advanced or Metastatic RET Fusion-Positive Solid Tumors. 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-solid-tumors (accessed on 9 May 2024).
- EMA. European Medicines Agency. Retsevmo. 2024. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/retsevmo (accessed on 5 September 2024).
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Lazurko, C.; Linder, R.; Pulman, K.; Lennox, G.; Feigenberg, T.; Fazelzad, R.; May, T.; Zigras, T. Bevacizumab Treatment for Low-Grade Serous Ovarian Cancer: A Systematic Review. Curr. Oncol. 2023, 30, 8159–8171. [Google Scholar] [CrossRef]
- Pfisterer, J.; Shannon, C.M.; Baumann, K.; Rau, J.; Harter, P.; Joly, F.; Sehouli, J.; Canzler, U.; Schmalfeldt, B.; Dean, A.P.; et al. Bevacizumab and platinum-based combinations for recurrent ovarian cancer: A randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Kubo-Kaneda, M.; Sunada, K.; Teishikata, Y.; Kitamura, A.; Okamoto, K.; Toriyabe, K.; Nii, M.; Yoshida, K.; Kondo, E.; et al. Adverse Events Associated With Long-term Treatment of Epithelial Ovarian Cancer With Bevacizumab and Chemotherapy. Anticancer Res. 2022, 42, 4165–4171. [Google Scholar] [CrossRef] [PubMed]
- Babaier, A.; Ghatage, P. Among patients with advanced ovarian carcinoma, who benefits from bevacizumab the most? Ann. Transl. Med. 2023, 11, 367. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Petrillo, M.; Paris, I.; Vizzielli, G.; Amadio, G.; Cosentino, F.; Salutari, V.; Scambia, G.; Fagotti, A. Neoadjuvant Chemotherapy Followed by Maintenance Therapy With or Without Bevacizumab in Unresectable High-Grade Serous Ovarian Cancer: A Case-Control Study. Ann. Surg. Oncol. 2015, 22, 952–958. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Gladieff, L.; Tholander, B.; Stroyakovsky, D.; Gore, M.; Scambia, G.; Kovalenko, N.; Oaknin, A.; Ronco, J.P.; Freudensprung, U.; et al. Efficacy and safety results from OCTAVIA, a single-arm phase II study evaluating front-line bevacizumab, carboplatin and weekly paclitaxel for ovarian cancer. Eur. J. Cancer 2013, 49, 3831–3838. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lim, M.C.; Kim, B.G.; Ngoi, N.Y.L.; Choi, C.H.; Park, S.Y.; Tan, D.S.P.; Go, Y.; Lee, J.Y. A single-arm phase ii study of olaparib maintenance with pembrolizumab and bevacizumab in brca non-mutated patients with platinum-sensitive recurrent ovarian cancer (Opeb-01). J. Gynecol. Oncol. 2021, 32, e31. [Google Scholar] [CrossRef]
- Kim, Y.N.; Park, B.; Kim, J.W.; Kim, B.G.; Kim, S.W.; Kim, H.S.; Choi, C.H.; Lim, M.C.; Ngoi, N.; Tan, D.; et al. Triplet maintenance therapy of olaparib, pembrolizumab and bevacizumab in women with BRCA wild-type, platinum-sensitive recurrent ovarian cancer: The multicenter, single-arm phase II study OPEB-01/APGOT-OV4. Nat. Commun. 2023, 14, 5476. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Guo, J.; Wei, P. Direct antitumor activity of bevacizumab: An overlooked mechanism? Front. Pharmacol. 2024, 15, 1394878. [Google Scholar] [CrossRef]
- Newtson, A.; Reyes, H.; Devor, E.J.; Goodheart, M.J.; Bosquet, J.G. Identification of novel fusion transcripts in high grade serous ovarian cancer. Int. J. Mol. Sci. 2021, 22, 4791. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Li, Z.; Xie, F.; Pang, Y.; Zhang, C.; Tang, H.; Zhang, H.; Chen, C.; Zhan, Y.; Zhao, T.; et al. Oncogenic and drug-sensitive RET mutations in human epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2020, 39, 53. [Google Scholar] [CrossRef]
- Rushton, T.; Krause, H.B.; Samec, T.; Elliott, A.; Karnezis, A.N.; Toboni, M.D.; Thaker, P.H.; Braxton, D.R.; Oberley, M.; Gershenson, D.M.; et al. Characterizing the genomic landscape through the lens of FOLR1 status in low and high grade serous ovarian carcinoma. Gynecol. Oncol. 2024, 191, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, G.; Nagasaka, M. Oncogenic fusions: Targeting NTRK. Crit. Rev. Oncol. Hematol. 2024, 194, 104234. [Google Scholar] [CrossRef]
- Penault-Llorca, F.; Rudzinski, E.R.; Sepulveda, A.R. Testing algorithm for identification of patients with TRK fusion cancer. J. Clin. Pathol. 2019, 72, 460–467. [Google Scholar] [CrossRef]
- Cocco, E.; Benhamida, J.; Middha, S.; Zehir, A.; Mullaney, K.; Shia, J.; Yaeger, R.; Zhang, L.; Wong, D.; Villafania, L.; et al. Colorectal carcinomas containing hypermethylated MLH1 promoter and wild-type BRAF/KRAS are enriched for targetable kinase fusions. Cancer Res. 2019, 79, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Shen, L.; Hong, D.S.; McDermott, R.; Keedy, V.L.; Casanova, M.; Demetri, G.D.; Dowlati, A.; Melcón, S.G.; Lassen, U.N.; et al. Larotrectinib efficacy and safety in adult patients with tropomyosin receptor kinase fusion sarcomas. Cancer 2023, 129, 3772–3782. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Kotnik, E.N.; Mullen, M.M.; Spies, N.C.; Li, T.; Inkman, M.; Zhang, J.; Martins-Rodrigues, F.; Hagemann, I.S.; McCourt, C.K.; Thaker, P.H.; et al. Genetic characterization of primary and metastatic high-grade serous ovarian cancer tumors reveals distinct features associated with survival. Commun. Biol. 2023, 6, 688. [Google Scholar] [CrossRef]
- Federman, N.; McDermott, R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 931–939. [Google Scholar] [CrossRef]
- Scott, L.J. Larotrectinib: First Global Approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Carlson, J.J.; Italiano, A.; Brose, M.S.; Federman, N.; Lassen, U.; Kummar, S.; Sullivan, S.D. Comparative Effectivness of Larotrectinib and Entrectinib for TRK Fusion Cancer. Am. J. Manag. Care 2022, 28 (Suppl. S2), S26. Available online: www.ajmc.com (accessed on 13 November 2024).
- Li, Y.; Gao, Y.; Zhang, X.; Guo, H.; Gao, H. Nanoparticles in precision medicine for ovarian cancer: From chemotherapy to immunotherapy. Int. J. Pharm. 2020, 591, 119986. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.B. Larotrectinib and Entrectinib: TRK Inhibitors for the Treatment of Pediatric and Adult Patients with NTRK Gene Fusion. J. Adv. Pract. Oncol. 2020, 11, 418. [Google Scholar]
- Al-Salama, Z.T.; Keam, S.J. Entrectinib: First Global Approval. Drugs 2019, 79, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Subbiah, V.; Yang, D.; Vamsidhar, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Belli, C.; Anand, S.; Gainor, J.F.; Penault-Llorca, F.; Subbiah, V.; Drilon, A.; Andre, F.; Curigliano, G. Progresses toward precision medicine in RET-altered solid tumors. Clin. Cancer Res. 2020, 26, 6102–6111. [Google Scholar] [CrossRef]
- Markham, A. Selpercatinib: First Approval. Drugs 2020, 80, 1119–1124. [Google Scholar] [CrossRef]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. Lancet Oncol. 2022, 23, 1261–1273. [Google Scholar] [CrossRef]
- Duke, E.S.; Bradford, D.; Marcovitz, M.; Amatya, A.K.; Mishra-Kalyani, P.S.; Nguyen, E.; Price, L.S.L.; Zirkelbach, J.F.; Li, Y.; Bi, Y.; et al. FDA Approval Summary: Selpercatinib for the Treatment of Advanced RET Fusion-Positive Solid Tumors. Clin. Cancer Res. 2023, 29, 3573–3578. [Google Scholar] [CrossRef] [PubMed]
- Chavda, J.; Bhatt, H. Systemic review on B-RafV600E mutation as potential therapeutic target for the treatment of cancer. Eur. J. Med. Chem. 2020, 206, 112675. [Google Scholar] [CrossRef] [PubMed]
- Bösmüller, H.; Fischer, A.; Pham, D.L.; Fehm, T.; Capper, D.; Von Deimling, A.; Bonzheim, I.; Staebler, A.; Fend, F. Detection of the BRAF V600E mutation in serous ovarian tumors: A comparative analysis of immunohistochemistry with a mutation-specific monoclonal antibody and allele-specific PCR. Hum. Pathol. 2013, 44, 329–335. [Google Scholar] [CrossRef]
- White, R.; Otaibi, Z.; Rao, R.; Finley, G. BRAF V600E Mutation in Multiple Primary Malignancies: A Hairy Affair. Cureus 2018, 10, e3600. [Google Scholar] [CrossRef]
- Maji, L.; Teli, G.; Raghavendra, N.M.; Sengupta, S.; Pal, R.; Ghara, A.; Matada, G.S.P. An updated literature on BRAF inhibitors (2018–2023). Mol. Divers. 2023, 28, 2689–2730. [Google Scholar] [CrossRef]
- Chui, M.H.; Chang, J.C.; Zhang, Y.; Zehir, A.; Schram, A.M.; Konner, J.; Drilon, A.E.; Da, A.; Paula, C.; Weigelt, B.; et al. Spectrum of BRAF Mutations and Gene Rearrangements in Ovarian Serous Carcinoma. JCO Precis. Oncol. 2021, 5, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.E.; Roszik, J.; Janku, F.; Hong, D.S.; Kato, S.; Naing, A.; Piha-Paul, S.; Fu, S.; Tsimberidou, A.; Cabanillas, M.; et al. BRAF v600E–mutant cancers treated with vemurafenib alone or in combination with everolimus, sorafenib, or crizotinib or with paclitaxel and carboplatin (VEM-PLUS) study. NPJ Precis. Oncol. 2023, 7, 19. [Google Scholar] [CrossRef]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, J.; Mcshane, L.M.; Rubinstein, V.; et al. Dabrafenib and Trametinib in Patients with Tumors with BRAF V600E Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: Updated analysis from the phase II ROAR basket study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef]
- Heo, Y.A. Mirvetuximab Soravtansine: First Approval. Drugs 2023, 83, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, M.C. Introduction to antibody-drug conjugates. Antibodies 2021, 10, 42. [Google Scholar] [CrossRef]
- Manzano, A.; Ocaña, A. Antibody-drug conjugates: A promising novel therapy for the treatment of ovarian cancer. Cancers 2020, 12, 2223. [Google Scholar] [CrossRef]
- Anastasio, M.K.; Shuey, S.; Davidson, B.A. Antibody-Drug Conjugates in Gynecologic Cancers. Curr. Treat. Options Oncol. 2024, 25, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.M.; Eskander, R.N.; Binder, P.S. Recent Therapeutic Advances in Gynecologic Oncology: A Review. Cancers 2024, 16, 770. [Google Scholar] [CrossRef]
- Stewart, D.; Cristea, M. Antibody-drug conjugates for ovarian cancer: Current clinical development. Curr. Opin. Obstet. Gynecol. 2019, 31, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ochoa, E.; Veneziani, A.C.; Oza, A.M. Mirvetuximab Soravtansine in Platinum-Resistant Ovarian Cancer. Clin. Med. Insights Oncol. 2023, 17, 11795549231187264. [Google Scholar] [CrossRef]
- Kokori, E.; Olatunji, G.; Komolafe, R.; Abraham, I.C.; Ukoaka, B.; Samuel, O.; Ayodeji, A.; Ogunbowale, I.; Ezenwoba, C.; Aderinto, N. Mirvetuximab soravtansine: A breakthrough in targeted therapy for platinum-resistant ovarian cancer. Medicine 2024, 103, e38132. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Lorusso, D.; Oaknin, A.; Pignata, S.; Dean, A.; Denys, H.; Colombo, N.; Gorp, V.; Konner, J.A.; Marin, R.; et al. Efficacy and Safety of Mirvetuximab Soravtansine in Patients with Platinum-Resistant Ovarian Cancer with High Folate Receptor Alpha Expression: Results From the SORAYA Study. J. Clin. Oncol. 2023, 41, 2436–2445. [Google Scholar] [CrossRef]
- Zhu, F.; Tu, Y.P.; Sloss, C.; Wang, Y. Assessment of mirvetuximab soravtansine immunogenicity in patients with folate receptor alpha-positive ovarian cancer. Bioanalysis 2024, 16, 1101–1113. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Oaknin, A.; Cecere, S.C.; Denys, H.; Colombo, N.; van Gorp, T.; Konner, J.A.; Romeo Marin, M.; Harter, P.; et al. Mirvetuximab soravtansine in folate receptor alpha (FRα)–high platinum-resistant ovarian cancer: Final overall survival and post hoc sequence of therapy subgroup results from the SORAYA trial. Int. J. Gynecol. Cancer 2024, 34, 1119. Available online: https://ijgc.bmj.com/lookup/doi/10.1136/ijgc-2024-005401 (accessed on 9 September 2024). [CrossRef]
- Richardson, D.L.; Moore, K.N.; Vergote, I.; Gilbert, L.; Martin, L.P.; Mantia-Smaldone, G.M.; Castro, C.M.; Provencher, D.; Matulonis, U.A.; Stec, J.; et al. Phase 1b study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate, in combination with carboplatin and bevacizumab in patients with platinum-sensitive ovarian cancer. Gynecol. Oncol. 2024, 185, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Oza, A.M.; Colombo, N.; Oaknin, A.; Scambia, G.; Lorusso, D.; Konecny, G.E.; Banerjee, S.; Murphy, C.G.; Tanyi, J.L.; et al. Phase III, randomized trial of mirvetuximab soravtansine versus chemotherapy in patients with platinum-resistant ovarian cancer: Primary analysis of FORWARD I. Ann. Oncol. 2021, 32, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, K.; Barton, W.; Wheeler, L.; Smith, H.J.; Mythreye, K.; Arend, R.C. Targeted therapy in high grade serous ovarian Cancer: A literature review. Gynecol. Oncol. Rep. 2024, 54, 101450. [Google Scholar] [CrossRef]
- Arend, R.C.; Jackson-Fisher, A.; Jacobs, I.A.; Chou, J.; Monk, B.J. Ovarian cancer: New strategies and emerging targets for the treatment of patients with advanced disease. Cancer Biol. Ther. 2021, 22, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Appleton, K.M.; Elrod, A.K.; Lassahn, K.A.; Shuford, S.; Holmes, L.M.; DesRochers, T.M. PD-1/PD-L1 checkpoint inhibitors in combination with olaparib display antitumor activity in ovarian cancer patient-derived three-dimensional spheroid cultures. Cancer Immunol. Immunother. 2021, 70, 843–856. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Wenham, R.M.; Wahner-Hendrickson, A.; Armstrong, D.K.; Chan, N.; O’Malley, D.M.; Lee, J.M.; Penson, R.T.; Cristea, M.C.; et al. A phase 2 trial exploring the significance of homologous recombination status in patients with platinum sensitive or platinum resistant relapsed ovarian cancer receiving combination cediranib and olaparib. Gynecol. Oncol. 2024, 187, 105–112. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0090825824002142 (accessed on 13 November 2024). [CrossRef]
- Park, J.; Kim, J.C.; Lee, M.; Lee, J.H.; Kim, Y.N.; Lee, Y.J.; Kim, S.; Kim, S.W.; Park, S.H.; Lee, J.Y. Frequency of peripheral PD-1+regulatory T cells is associated with treatment responses to PARP inhibitor maintenance in patients with epithelial ovarian cancer. Br. J. Cancer 2023, 129, 1841–1851. [Google Scholar] [CrossRef]
- Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Maiello, E. Ovarian cancer in the era of immune checkpoint inhibitors: State of the art and future perspectives. Cancers 2021, 13, 4438. [Google Scholar] [CrossRef]
- Wang, Q.; Lou, W.; Di, W.; Wu, X. Prognostic value of tumor PD-L1 expression combined with CD8+ tumor infiltrating lymphocytes in high grade serous ovarian cancer. Int. Immunopharmacol. 2017, 52, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Wu, H.; Wu, J.; Ding, P.; He, J.; Sang, M.; Liu, L. Mechanisms of immune checkpoint inhibitors: Insights into the regulation of circular RNAS involved in cancer hallmarks. Cell Death Dis. 2024, 15, 3. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Qvortrup, C.; Hansen, K.H. How to select cancer patients for immunotherapy. eBioMedicine 2021, 63, 103184. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Vergote, L.B.; Sehouli, J.; Salutari, V.; Zola, P.; Madry, R.; Wenham, R.M.; Korach, J.; Pautier, P.; Cibula, D.; et al. ENGOT-ov43/KEYLYNK-001: A phase III trial of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA’-nonmutated advanced epithelial ovarian cancer. Ann. Oncol. 2019, 30, ix77. [Google Scholar] [CrossRef]
- Cheng, X.; Li, P.; Jiang, R.; Meng, E.; Wu, H. ADC: A deadly killer of platinum resistant ovarian cancer. J. Ovarian Res. 2024, 17, 196. [Google Scholar] [CrossRef] [PubMed]
- Atallah, G.A.; Kampan, N.C.; Chew, K.T.; Mohd Mokhtar, N.; Md Zin, R.R.; Shafiee, M.N.B.; Abd. Aziz, N.H.B. Predicting Prognosis and Platinum Resistance in Ovarian Cancer: Role of Immunohistochemistry Biomarkers. Int. J. Mol. Sci. 2023, 24, 1973. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wan, Y.; Zhang, L.; Meng, H.; Yuan, L.; Zhou, S.; Cheng, W.; Jiang, Y. Beyond monotherapy: An era ushering in combinations of PARP inhibitors with immune checkpoint inhibitors for solid tumors. Biomed. Pharmacother. 2024, 175, 116733. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-ov45): A randomized, phase III trial to evaluate rucaparib as monotherapy (ATHENA–MONO) and rucaparib in combination with nivolumab (ATHENA–COMBO) as maintenance treatment following frontline platinum-based chemotherapy in ovarian cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar]
- Ali, E.; Ellahi, A.; Adil, M.; Shaikh, A.; Huda, Z. Jemperli (Dostarlimab-gxly): An unprecedented cancer trial. Ann. Med. Surg. 2022, 79, 104047. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, L.; Han, L.; Zheng, X.; Tong, R.; Li, L.; Bai, L.; Bian, Y. Immune-related adverse events of immune checkpoint inhibitors: A review. Front. Immunol. 2023, 14, 1167975. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Gonzalez-Martin, A.; Cruz, F.M.; Friedlander, M.; Glasspool, R.; Lorusso, D.; Marth, C.; Monk, B.J.; Kim, J.W.; Hinson, P.; et al. EPIK-O/ENGOT-OV61: Alpelisib plus olaparib vs cytotoxic chemotherapy in high-grade serous ovarian cancer (phase III study). Future Oncol. 2022, 18, 3481–3492. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Ye, Y.; Huang, Z.; Zhang, M.; Li, J.; Zhang, Y.; Lou, C. Synergistic therapeutic potential of alpelisib in cancers (excluding breast cancer): Preclinical and clinical evidences. Biomed. Pharmacother. 2023, 159, 114183. [Google Scholar] [CrossRef]
- Bauer, T.M.; Moore, K.N.; Rader, J.S.; Simpkins, F.; Mita, A.C.; Beck, J.T.; Hart, L.; Chu, Q.; Oza, A.; Tinker, A.V.; et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target Oncol. 2023, 18, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Sachdev, J.; Imedio, E.R.; Kumar, S.; Mugundu, G.M.; Jenkins, S.; Chmielecki, J.; Jones, S.; Spigel, D.R.; Johnson, M. A phase Ib study of adavosertib, a selective Wee1 inhibitor, in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2023, 41, 493–502. [Google Scholar] [CrossRef]
- Embaby, A.; Kutzera, J.; Geenen, J.J.; Pluim, D.; Hofland, I.; Sanders, J.; Lopez-Yurda, M.; Beijnen, J.H.; Huitema, A.D.R.; Witteveen, P.O.; et al. WEE1 inhibitor adavosertib in combination with carboplatin in advanced TP53 mutated ovarian cancer: A biomarker-enriched phase II study. Gynecol. Oncol. 2023, 174, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Roering, P.; Siddiqui, A.; Heuser, V.D.; Potdar, S.; Mikkonen, P.; Oikkonen, J.; Li, Y.; Pikkusaari, S.; Wennerberg, K.; Hynninen, J.; et al. Effects of Wee1 inhibitor adavosertib on patient-derived high-grade serous ovarian cancer cells are multiple and independent of homologous recombination status. Front. Oncol. 2022, 12, 954430. [Google Scholar] [CrossRef]
- Leijen, S.; Van Geel, R.M.J.M.; Sonke, G.S.; De Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; Van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patientswith tp53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin. Cancer Res. 2022, 28, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef] [PubMed]
- Biegała, Ł.; Gajek, A.; Szymczak-Pajor, I.; Marczak, A.; Śliwińska, A.; Rogalska, A. Targeted inhibition of the ATR/CHK1 pathway overcomes resistance to olaparib and dysregulates DNA damage response protein expression in BRCA2 MUT ovarian cancer cells. Sci. Rep. 2023, 13, 22659. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.; Tanyi, J.L.; Latif, N.; Morgan, M.; Torigian, D.A.; Rodriguez, D.; Smithm, S.A.; Dean, E.; et al. Combination ATR (ceralasertib) and PARP (olaparib) Inhibitor (CAPRI) Trial in Acquired PARP Inhibitor–Resistant Homologous Recombination–Deficient Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2800–2807. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; da Costa, A.A.B.A.; Gulhan, D.; Lee, E.K.; Cheng, S.C.; Hendrickson, A.E.W.; Kochupurakkal, B.; Kolin, D.L.; Kohn, E.C.; Liu, J.F.; et al. A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat. Commun. 2021, 12, 5574. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Moore, R.G.; Ghamande, S.; Park, M.S.; Diaz, J.P.; Chapman, J.; Kendrick, J.; Slomovitz, B.M.; Tewari, K.S.; Lowe, E.S.; et al. Cediranib in Combination with Olaparib in Patients without a Germline BRCA1/2 Mutation and with Recurrent Platinum-Resistant Ovarian Cancer: Phase IIb CONCERTO Trial. Clin. Cancer Res. 2022, 28, 4186–4193. [Google Scholar] [CrossRef]
- Passaro, A.; Al Bakir, M.; Hamilton, E.G.; Diehn, M.; André, F.; Roy-Chowdhuri, S.; Mountzios, G.; Wistuba, I.I.; Swanton, C.; Peters, S. Cancer biomarkers: Emerging trends and clinical implications for personalized treatment. Cell 2024, 187, 1617–1635. [Google Scholar] [CrossRef]
- Bodaghi, A.; Fattahi, N.; Ramazani, A. Biomarkers: Promising and valuable tools towards diagnosis, prognosis and treatment of COVID-19 and other diseases. Heliyon 2023, 9, e13323. [Google Scholar] [CrossRef]
- Sechidis, K.; Papangelou, K.; Metcalfe, P.D.; Svensson, D.; Weatherall, J.; Brown, G. Distinguishing prognostic and predictive biomarkers: An information theoretic approach. Bioinformatics 2018, 34, 3365–3376. [Google Scholar] [CrossRef]
- Rodrigues-Ferreira, S.; Nahmias, C. Predictive biomarkers for personalized medicine in breast cancer. Cancer Lett. 2022, 545, 215828. [Google Scholar] [CrossRef] [PubMed]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Holý, P.; Hlaváč, V.; Šeborová, K.; Šůsová, S.; Tesařová, T.; Rob, L.; Hruda, M.; Bouda, J.; Bartáková, A.; Mrhalová, M.; et al. Targeted DNA sequencing of high-grade serous ovarian carcinoma reveals association of TP53 mutations with platinum resistance when combined with gene expression. Int. J. Cancer 2024, 155, 104–116. [Google Scholar] [CrossRef]
- Havasi, A.; Cainap, S.S.; Havasi, A.T.; Cainap, C. Ovarian Cancer—Insights into Platinum Resistance and Overcoming It. Medicina 2023, 59, 544. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, W.R.; Beach, J.A.; Karlan, B.Y. Reversing platinum resistance in high-grade serous ovarian carcinoma: Targeting BRCA and the homologous recombination system. Front. Oncol. 2014, 4, 34. [Google Scholar] [CrossRef]
- Grabska, K.; Pilarska, I.; Fudalej, M.M.; Deptała, A.; Badowska-Kozakiewicz, A. What is new about ovarian malignancies? Wspolczesna Onkol. 2021, 25, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Vikramdeo, K.S.; Sudan, S.K.; Singh, S.; Wilhite, A.; Dasgupta, S.; Rocconi, R.P.; Singh, A.P. Platinum-resistant ovarian cancer: From drug resistance mechanisms to liquid biopsy-based biomarkers for disease management. Semin. Cancer Biol. 2021, 77, 99–109. [Google Scholar] [CrossRef]
- Al-Moghrabi, N.; Al-Showimi, M.; Alqahtani, A.; Almalik, O.; Alhusaini, H.; Almalki, G.; Saad, A.; Alsunayi, E. Constitutional BRCA1 and MGMT Methylation Are Significant Risk Factors for Triple-Negative Breast Cancer and High-Grade Serous Ovarian Cancer in Saudi Women. Int. J. Mol. Sci. 2024, 25, 3108. [Google Scholar] [CrossRef]
- Flores-Colón, M.; Rivera-Serrano, M.; Reyes-Burgos, V.G.; Rolón, J.G.; Pérez-Santiago, J.; Marcos-Martínez, M.J.; Valiyeva, F.; Vivas-Mejía, P.E. MicroRNA Expression Profiles in Human Samples and Cell Lines Revealed Nine miRNAs Associated with Cisplatin Resistance in High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2024, 25, 3793. [Google Scholar] [CrossRef]
- Zhang, Q.; Ding, J.; Wang, Y.; He, L.; Xue, F. Tumor microenvironment manipulates chemoresistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 102. [Google Scholar] [CrossRef]
- Ardighieri, L.; Missale, F.; Bugatti, M.; Gatta, L.B.; Pezzali, I.; Monti, M.; Gottardi, S.; Zanotti, L.; Bignotti, E.; Ravaggi, A.; et al. Infiltration by CXCL10 Secreting Macrophages Is Associated with Antitumor Immunity and Response to Therapy in Ovarian Cancer Subtypes. Front. Immunol. 2021, 12, 690201. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, F.; Jiang, X.; Zhang, W.; Xiang, T.; Pan, Q.; Cai, L.; Zhao, J.; Weng, D.; Li, Y.; et al. CircITGB6 promotes ovarian cancer cisplatin resistance by resetting tumor-associated macrophage polarization toward the M2 phenotype. J. Immunother. Cancer 2022, 10, e004029. [Google Scholar] [CrossRef] [PubMed]
- Yang-Hartwich, Y.; Soteras, M.G.; Lin, Z.P.; Holmberg, J.; Sumi, N.; Craveiro, V.; Liang, M.; Romanoff, E.; Bingham, J.; Garofalo, F.; et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene 2015, 34, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- Oturkar, C.C.; Gandhi, N.; Rao, P.; Eng, K.H.; Miller, A.; Singh, P.K.; Zsiros, E.; Odunsi, K.O.; Das, G.M. Estrogen Receptor-beta2 (ERβ2)–Mutant p53–FOXM1 Axis: A Novel Driver of Proliferation, Chemoresistance, and Disease Progression in High Grade Serous Ovarian Cancer (HGSOC). Cancers 2022, 14, 1120. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Rodrigues, M.; Sandoval, J.L.; Kurtz, J.E.; Heitz, F.; Mosconi, A.M.; Romero, I.; Denison, U.; Nagao, S.; Vergote, I.; et al. Association of location of BRCA1 and BRCA2 mutations with benefit from olaparib and bevacizumab maintenance in high-grade ovarian cancer: Phase III PAOLA-1/ENGOT-ov25 trial subgroup exploratory analysis. Ann. Oncol. 2023, 34, 152–162. [Google Scholar] [CrossRef]
- Yubero, A.; Barquín, A.; Estévez, P.; Pajares, B.; Sánchez, L.; Reche, P.; Alarcón, J.; Calzas, J.; Gaba, L.; Fuentes, J.; et al. Rucaparib in recurrent ovarian cancer: Real-world experience from the rucaparib early access programme in Spain—A GEICO study. BMC Cancer 2022, 22, 1150. [Google Scholar] [CrossRef]
- Monk, B.J.; González-Martin, A.; Buckley, L.; Matulonis, U.A.; Rimel, B.J.; Wu, X.; Moore, K.N.; Mirza, M.R. Safety and management of niraparib monotherapy in ovarian cancer clinical trials. Int. J. Gynecol. Cancer 2023, 33, 971–981. [Google Scholar] [CrossRef]
- Frenel, J.S.; Kim, J.W.; Aryal, N.; Asher, R.; Berton, D.; Vidal, L.; Pautier, P.; Ledermann, J.A.; Penson, R.T.; Oza, A.M.; et al. Efficacy of subsequent chemotherapy for patients with BRCA1/2-mutated recurrent epithelial ovarian cancer progressing on olaparib versus placebo maintenance: Post-hoc analyses of the SOLO2/ENGOT Ov-21 trial. Ann. Oncol. 2022, 33, 1021–1028. [Google Scholar] [CrossRef]
- Disilvestro, P.; Banerjee, S.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Overall Survival with Maintenance Olaparib at a 7-Year Follow-Up in Patients with Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation: The SOLO1/GOG 3004 Trial. J. Clin. Oncol. 2022, 41, 609–617. [Google Scholar] [CrossRef]
- Cecere, S.C.; Giannone, G.; Salutari, V.; Arenare, L.; Lorusso, D.; Ronzino, G.; Lauria, R.; Cormio, G.; Carella, C.; Scollo, P.; et al. Olaparib as maintenance therapy in patients with BRCA 1–2 mutated recurrent platinum sensitive ovarian cancer: Real world data and post progression outcome. Gynecol. Oncol. 2020, 156, 38–44. [Google Scholar] [CrossRef]
- Smith, M.; Pothuri, B. Appropriate Selection of PARP Inhibitors in Ovarian Cancer. Curr. Treat. Options Oncol. 2022, 23, 887–903. [Google Scholar] [CrossRef]
- Lorusso, D.; Mouret-Reynier, M.A.; Harter, P.; Cropet, C.; Caballero, C.; Wolfrum-Ristau, P.; Satoh, T.; Vergote, I.; Parma, G.; Nøttrup, T.J.; et al. Updated progression-free survival and final overall survival with maintenance olaparib plus bevacizumab according to clinical risk in patients with newly diagnosed advanced ovarian cancer in the phase III PAOLA-1/ENGOT-ov25 trial. Int. J. Gynecol. Cancer 2023, 34, 550–558. [Google Scholar] [CrossRef]
- Matuszczak, M.; Kiljańczyk, A.; Marciniak, W.; Derkacz, R.; Stempa, K.; Baszuk, P.; Bryśkiewicz, M.; Cybulski, C.; Dębniak, T.; Gronwald, J.; et al. Antioxidant Properties of Zinc and Copper—Blood Zinc-to Copper-Ratio as a Marker of Cancer Risk BRCA1 Mutation Carriers. Antioxidants 2024, 13, 841. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Sun, G. Targeting copper metabolism: A promising strategy for cancer treatment. Front. Pharmacol. 2023, 14, 1203447. [Google Scholar] [CrossRef] [PubMed]
- Lukanović, D.; Polajžer, S.; Matjašič, M.; Kobal, B.; Černe, K. Analysis of ATP7A Expression and Ceruloplasmin Levels as Biomarkers in Patients Undergoing Neoadjuvant Chemotherapy for Advanced High-Grade Serous Ovarian Carcinoma. Int. J. Mol. Sci. 2024, 25, 10195. [Google Scholar] [CrossRef]
- Kiljańczyk, A.; Matuszczak, M.; Marciniak, W.; Derkacz, R.; Stempa, K.; Baszuk, P.; Bryśkiewicz, M.; Lubiński, K.; Cybulski, C.; Dębniak, T.; et al. Blood Lead Level as Marker of Increased Risk of Ovarian Cancer in BRCA1 Carriers. Nutrients 2024, 16, 1370. [Google Scholar] [CrossRef] [PubMed]
- Kluza, M.; Paszek, S.; Kluza, K.; Januszek, S.; Potocka, N.; Skrzypa, M.; Zuchowska, A.; Wróbel, A.; Baszuk, P.; Marciniak, W.; et al. An Assessment of Serum Selenium Concentration in Women with Ovarian Cancer. Nutrients 2023, 15, 850. [Google Scholar] [CrossRef] [PubMed]
- Golara, A.; Kozłowski, M.; Guzik, P.; Kwiatkowski, S.; Cymbaluk-Płoska, A. The Role of Selenium and Manganese in the Formation, Diagnosis and Treatment of Cervical, Endometrial and Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 10887. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Wang, Y.; Duval, A.J.; Adli, M.; Matei, D. Biology-driven therapy advances in high-grade serous ovarian cancer. J. Clin. Investig. 2024, 134, e174013. [Google Scholar] [CrossRef]
- Suh, K.; Carlson, J.J.; Xia, F.; Williamson, T.; Sullivan, S.D. Comparative effectiveness of larotrectinib versus entrectinib for the treatment of metastatic NTRK gene fusion cancers. J. Comp. Eff. Res. 2022, 11, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Perrone, C.; Angioli, R.; Luvero, D.; Giannini, A.; Donato, V.; Di Cuccu, I.; Muzii, L.; Raspagliesi, F.; Bogani, G. Targeting BRAF pathway in low-grade serous ovarian cancer. J. Gynecol. Oncol. 2024, 35, e104. [Google Scholar] [CrossRef]
- De Braud, F.; Deschler-Baier, B.; Morris, J.C.; Worden, F.; Han, Y.; Kiiskinen, U.; Jen, M.H.; Barker, S.S.; Szymczak, S.; Gilligan, A.M. Comparative Effectiveness of First-Line Selpercatinib versus Standard Therapies in Patients with RET-Activated Cancers: An Exploratory Interpatient Analysis of LIBRETTO-001. Cancers 2024, 16, 140. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Wu, L.; Yang, L.; Sun, T.; Liu, X.; Yin, R.; Jiang, Y.; Li, J.; Li, Q. Therapeutic strategies targeting folate receptor α for ovarian cancer. Front. Immunol. 2023, 14, 1254532. [Google Scholar] [CrossRef]
- Ahnquist, J.; Wamala, S.P.; Lindstrom, M. Social determinants of health—A question of social or economic capital? Interaction effects of socioeconomic factors on health outcomes. Soc. Sci. Med. 2012, 74, 930–939. [Google Scholar] [CrossRef]
- Pilié, P.G.; George, A.; Yap, T.A. Patient selection biomarker strategies for PARP inhibitor therapy. Ann. Oncol. 2020, 31, 1603–1605. [Google Scholar] [CrossRef]
- Lourenção, M.; Simões Correa Galendi, J.; de Campos Reis Galvão, H.; Antoniazzi, A.P.; Grasel, R.S.; Carvalho, A.L.; Mauad, E.C.; de Oliveira, J.H.C.; Reis, R.M.; Mandrik, O.; et al. Cost-Effectiveness of BRCA 1/2 Genetic Test and Preventive Strategies: Using Real-World Data from an Upper-Middle Income Country. Front. Oncol. 2022, 12, 951310. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, C.; Lorusso, D.; Costa, F.; Armeni, P. Cost-Effectiveness Analysis of HRD Testing for Previously Treated Patients with Advanced Ovarian Cancer in Italy. Adv. Ther. 2024, 41, 1385–1400. [Google Scholar] [CrossRef]
- Gonzalez, R.; Havrilesky, L.J.; Myers, E.R.; Secord, A.A.; Dottino, J.A.; Berchuck, A.; Moss, H.A. Cost-effectiveness analysis comparing “PARP inhibitors-for-all” to the biomarker-directed use of PARP inhibitor maintenance therapy for newly diagnosed advanced stage ovarian cancer. Gynecol. Oncol. 2020, 159, 483–490. [Google Scholar] [CrossRef]
- Kim, J.B.; Kim, Y.I.; Yoon, Y.S.; Lee, J.L.; Kim, C.W.; Park, I.J.; Lim, S.B.; Yu, C.S.; Kim, J.C.; Kim, J.; et al. Cost-effective screening using a two-antibody panel for detecting mismatch repair deficiency in sporadic colorectal cancer. World J. Clin. Cases 2021, 9, 6999–7008. [Google Scholar] [CrossRef]
- Stephan, A.J.; Reuschenbach, M.; Saxena, K.; Prabhu, V.S.; Jacob, C.; Schneider, K.M.; Greiner, W.; Wölle, R.; Hampl, M. Healthcare Costs and Resource Use Associated with Cervical Intraepithelial Neoplasia and Cervical Conization: A Retrospective Study of German Statutory Health Insurance Claims Data. J. Health Econ. Outcomes Res. 2022, 9, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef]
- Li, W.Q.; Li, L.Y.; Bai, R.L.; Qian, L.; Chen, N.F.; Cui, J.W.; Wang, N.N. Cost-effectiveness of programmed cell death ligand 1 testing and tumor mutational burden testing of immune checkpoint inhibitors for advanced non-small cell lung cancer. Chin. Med. J. 2020, 133, 2630–2632. [Google Scholar] [CrossRef]
- CostHelper. Health. How Much Does Ovarian Cancer Treatment Cost? 2024. Available online: https://health.costhelper.com/ovarian-cancer.html#:~:text=For%20patients%20without%20health%20insurance%2C%20the%20cost%20of,treatment%20with%20surgery%20and%20chemotherapy%20for%20advanced%20cancer (accessed on 19 February 2025).
- Bercow, A.S.; Chen, L.; Chatterjee, S.; Tergas, A.I.; Hou, J.Y.; Burke, W.M.; Ananth, C.V.; Neugut, A.I.; Hershman, D.L.; Wright, J.D. Cost of care for the initial management of ovarian cancer. Obstet. Gynecol. 2017, 130, 1269–1275. [Google Scholar] [CrossRef]
- Goldsberry, W.N.; Summerlin, S.S.; Guyton, A.; Caddell, B.; Huh, W.K.; Kim, K.H.; Liang, M.I. The financial burden of PARP inhibitors on patients, payors, and financial assistance programs: Who bears the cost? Gynecol. Oncol. 2021, 160, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Dheer, P.; Kamerikar, V.; Fleming, M.; -Vazquez, S.; Gautam, R.; Lakhsmi, R.; Sharma, R.; Swami, S.; Willis, M.; Nilsson, K.; et al. EE284 Cost-Effectiveness Analysis of Larotrectinib and Entrectinib for Adult Patients with NTRK Gene Fusion-Positive Breast Cancer. Value Health 2024, 27, S110. [Google Scholar] [CrossRef]
- Vokinger, K.N.; Hwang, T.J.; Grischott, T.; Reichert, S.; Tibau, A.; Rosemann, T.; Kesselheim, A.S. Prices and clinical benefit of cancer drugs in the USA and Europe: A cost–benefit analysis. Lancet Oncol. 2020, 21, 664–670. [Google Scholar] [CrossRef]
- Yi, H.; Cao, Y.; Shi, F.; Wei, X.; Han, S. Cost-effectiveness analysis of selpercatinib versus chemotherapy and pembrolizumab in the first-line treatment of rearranged during transfection fusion–positive non-small cell lung cancer in the United States. Int. J. Clin. Pharm. 2024, 46, 1427–1435. [Google Scholar] [CrossRef]

| Medicine | Monotherapy | Combinations | Prior Response to | Special Considerations | References |
|---|---|---|---|---|---|
| Pt-Based Therapy | |||||
| (Complete or Partial) | |||||
| Primary Therapy | |||||
| bevacizumab | 1. carboplatin/ | N/A | 1. FIGO stages IIIB, IIIC, IV. | [17,18] | |
| paclitaxel | |||||
| Maintenance therapy following completion of primary Pt-based chemotherapy (CT) | |||||
| bevacizumab | yes | N/A | not specified | 1. FIGO stages IIIB, IIIC, IV. | [17,18] |
| olaparib | yes | N/A | yes | 1. BRCA1/2 mutation. | [19,20] |
| 2. FIGO stages III, IV. | |||||
| olaparib | N/A | bevacizumab | yes | 1. HRD-positive status. | [18,19] |
| 2. FIGO stages III, IV. | |||||
| 3. Following primary Pt-based CT in combination with bevacizumab. | |||||
| niraparib | yes | N/A | yes | 1. FIGO stages III, IV. | [21,22] |
| rucaparib | yes | N/A | yes | 1. FIGO stages III, IV. | [23,24] |
| FDA: not approved. | |||||
| Therapy of recurrent cancer | |||||
| bevacizumab | N/A | 1. carboplatin/ | yes | 1. First recurrence. | [17,18] |
| gemcitabin | 2. No prior anti-VEGF therapy. | ||||
| 2. carboplatin/ | |||||
| paclitaxel | |||||
| bevacizumab | N/A | 1. paclitaxel | no | 1. No more than 2 prior CT regimens. | [17,18] |
| 2. topotecan | 2. No prior anti-VEGF therapy. | ||||
| 3. doxorubicin PL | |||||
| Maintenance treatment of recurrent cancer | |||||
| bevacizumab | yes | N/A | not specified | Not specified | [17,18] |
| olaparib | yes | N/A | yes | 1. BRCA1/2-mutation. | [19,20] |
| niraparib | yes | N/A | yes | Not specified | [21,22] |
| FDA: germline BRCA-mutation. | |||||
| rucaparib | yes | yes | Not specified | [23,24] | |
| Patients who have no satisfactory treatment options | |||||
| larotrectinib | yes | N/A | no | 1. NTRK gene fusion. | [25,26] |
| entrectinib | yes | N/A | no | 1. NTRK gene fusion. | [27,28] |
| 2. No prior NTRK inhibitor therapy. | |||||
| selpercatinib | yes | N/A | no | 1. RET fusion. | [29,30] |
| dabrafenib + | N/A | N/A | no | 1. BRAF V600E mutation. | [31] |
| trametinib | FDA only approved. | ||||
| Pt-resistant tumour | |||||
| mirvetuximab | yes | N/A | no | 1. FRα positive. | [32,33] |
| sorvatansine | 2. One to three prior systemic treatment regimens. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polajžer, S.; Černe, K. Precision Medicine in High-Grade Serous Ovarian Cancer: Targeted Therapies and the Challenge of Chemoresistance. Int. J. Mol. Sci. 2025, 26, 2545. https://doi.org/10.3390/ijms26062545
Polajžer S, Černe K. Precision Medicine in High-Grade Serous Ovarian Cancer: Targeted Therapies and the Challenge of Chemoresistance. International Journal of Molecular Sciences. 2025; 26(6):2545. https://doi.org/10.3390/ijms26062545
Chicago/Turabian StylePolajžer, Sara, and Katarina Černe. 2025. "Precision Medicine in High-Grade Serous Ovarian Cancer: Targeted Therapies and the Challenge of Chemoresistance" International Journal of Molecular Sciences 26, no. 6: 2545. https://doi.org/10.3390/ijms26062545
APA StylePolajžer, S., & Černe, K. (2025). Precision Medicine in High-Grade Serous Ovarian Cancer: Targeted Therapies and the Challenge of Chemoresistance. International Journal of Molecular Sciences, 26(6), 2545. https://doi.org/10.3390/ijms26062545