
Establishing the Role of Liver Fatty Acid-Binding Protein in Post-Golgi Very-Low-Density Lipoprotein Trafficking Using a Novel Fluorescence-Based Assay



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
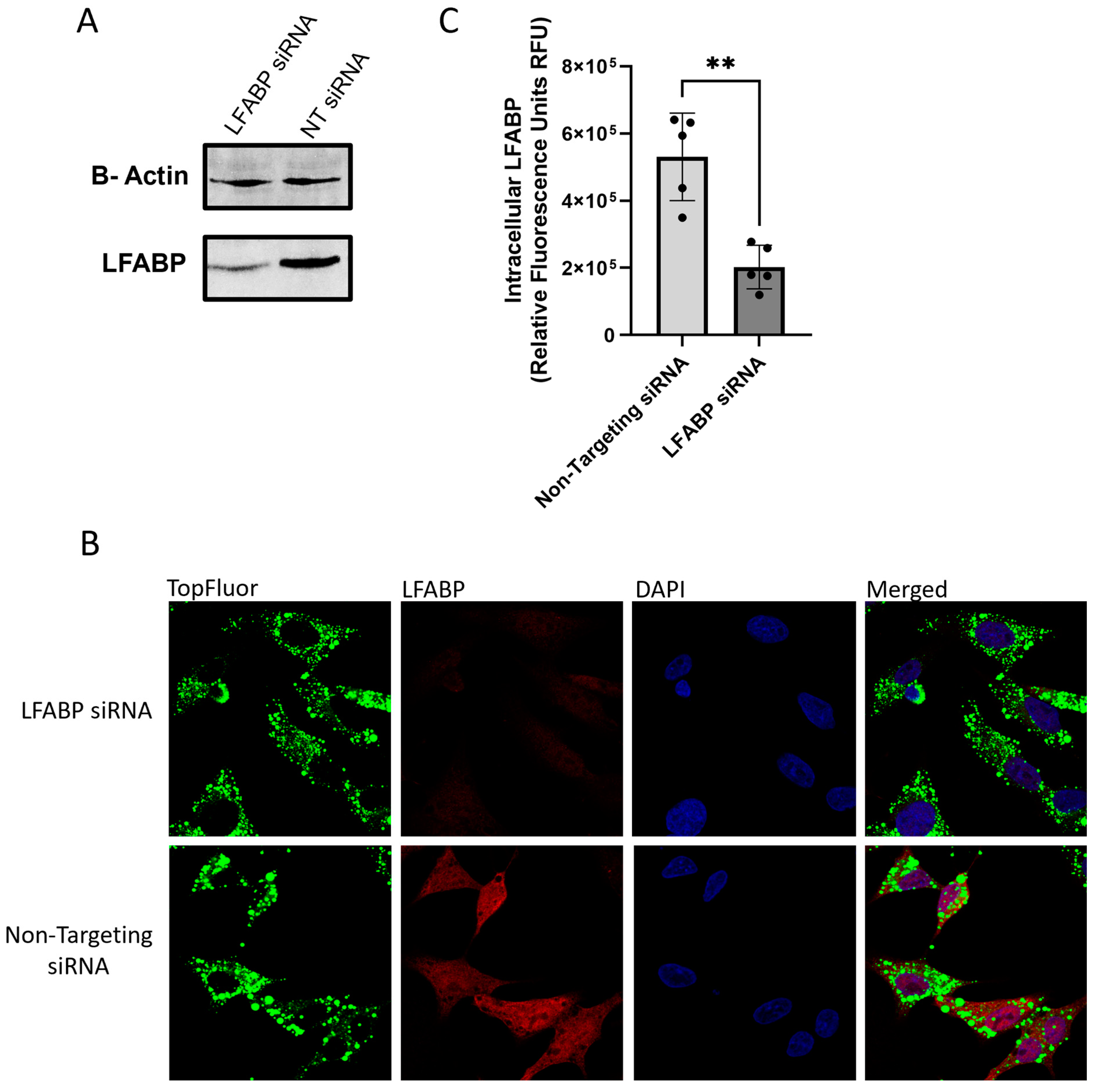
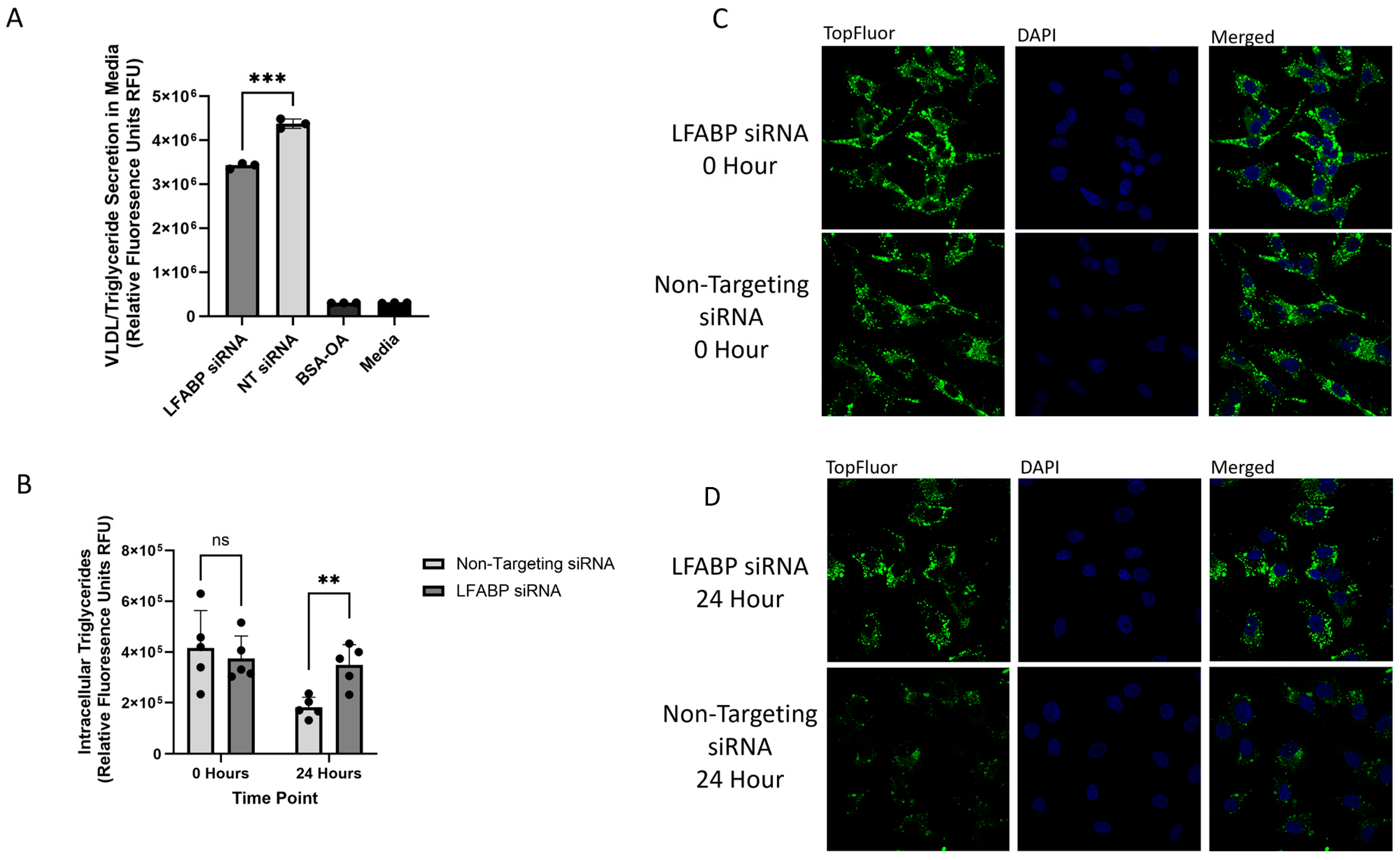
2.1. Measuring VLDL Secretion Through Fluorescence and ApoB100 Expression
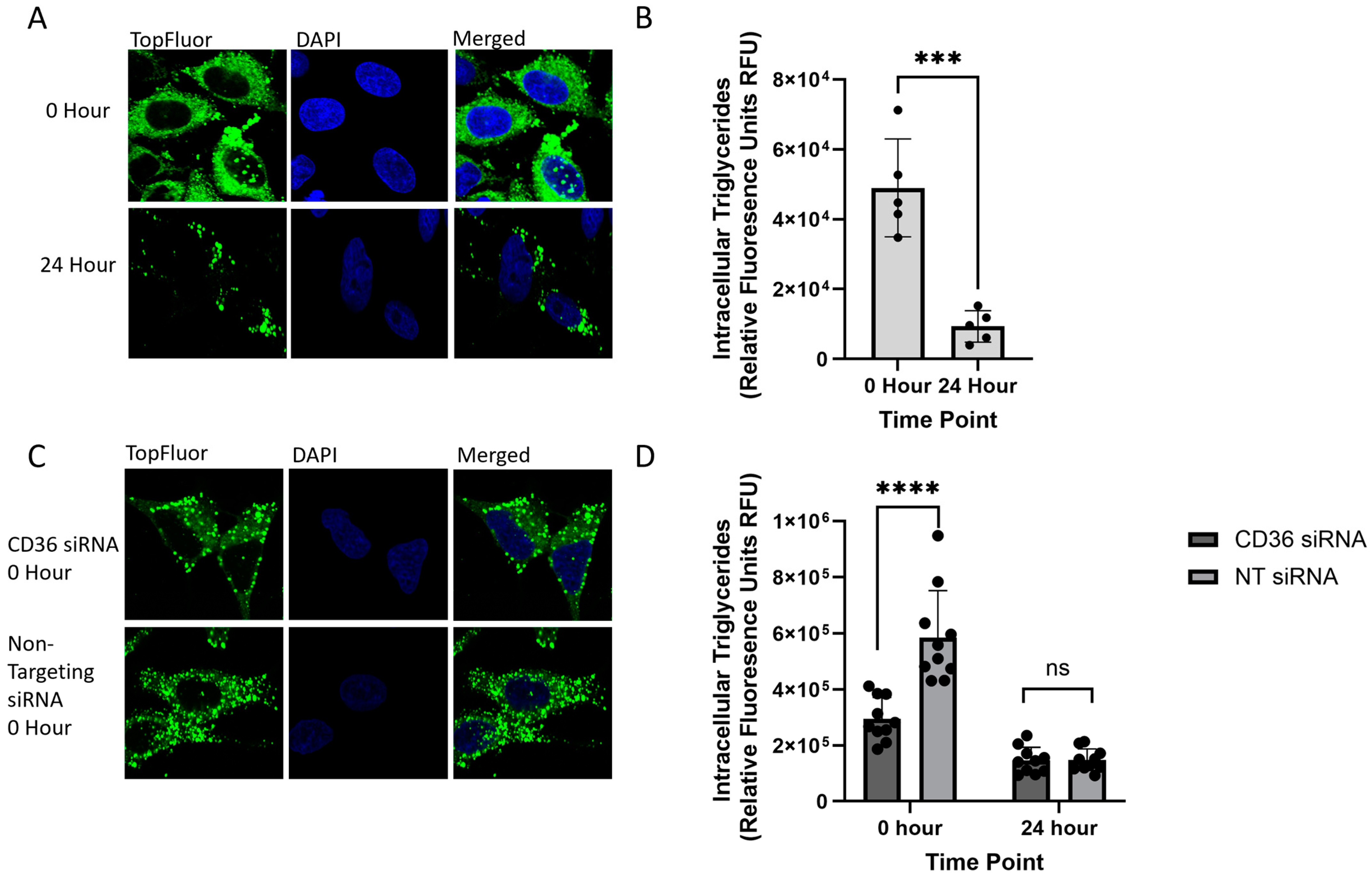
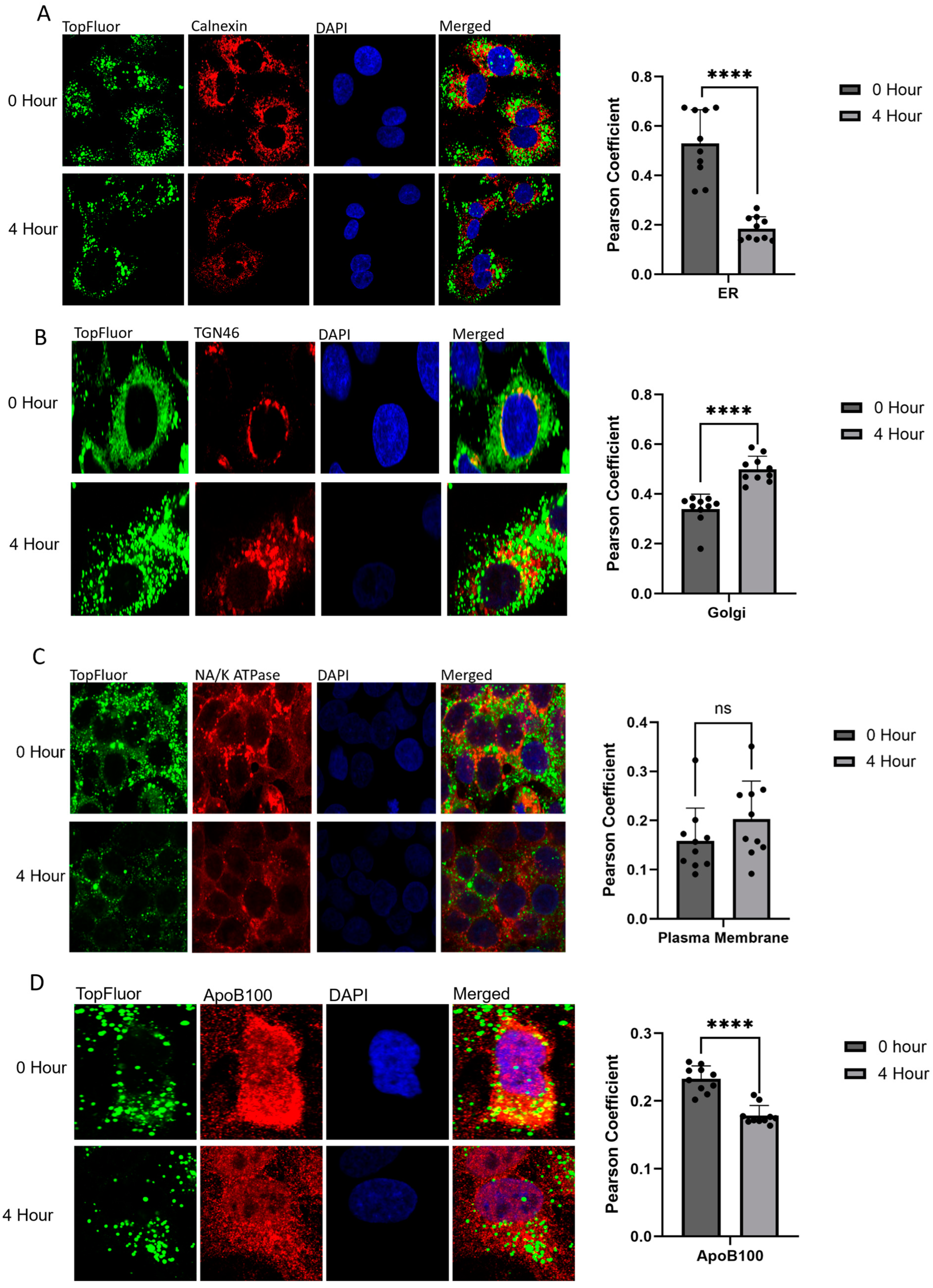
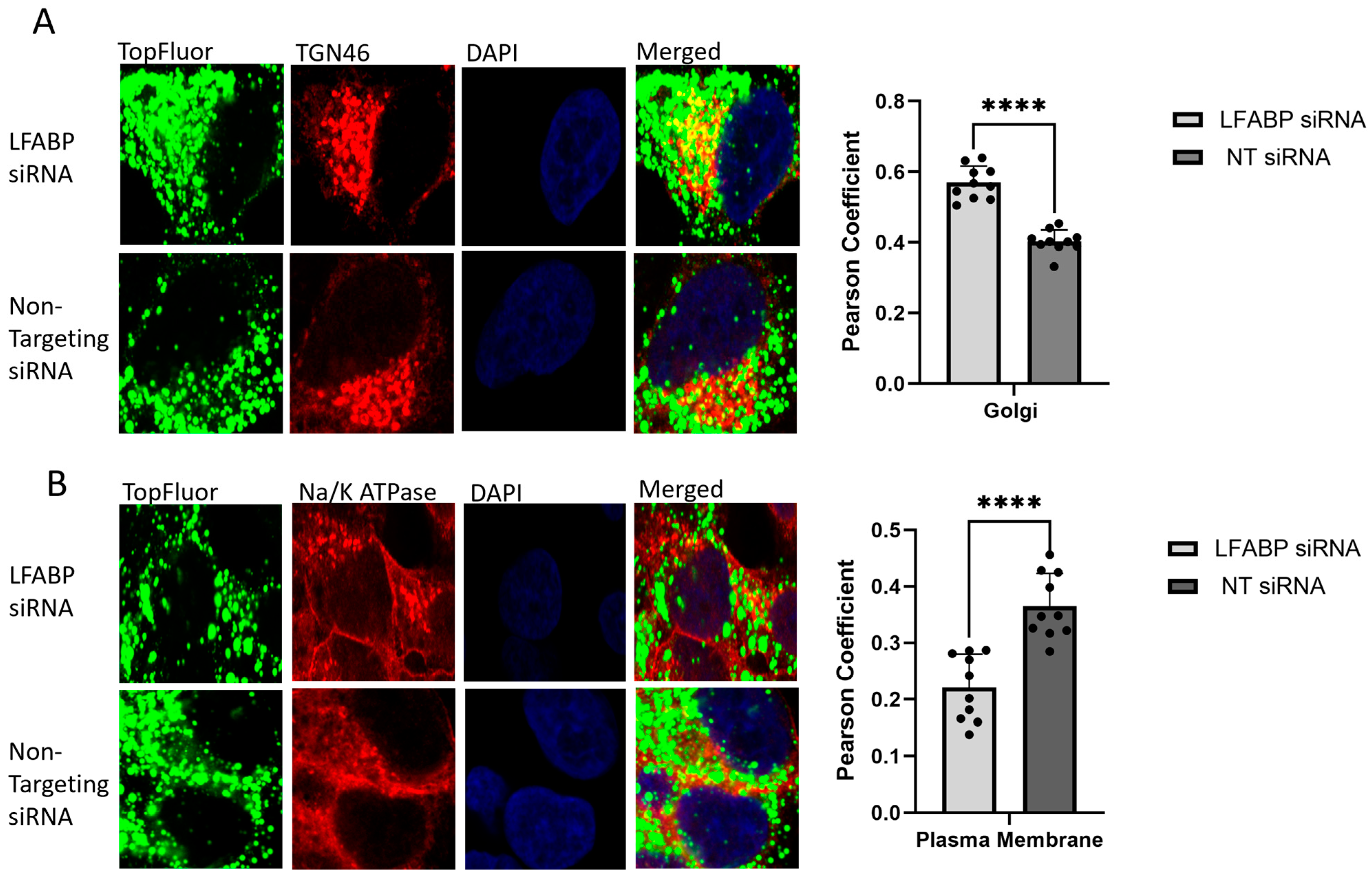
2.2. Monitoring Intracellular VLDL Trafficking Through Confocal Microscopy
2.3. Identifying Changes in the Rates of VLDL Secretion
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. TopFluor Treatment
4.3. Fluorescence Measurements
4.4. Lomitapide Treatment
4.5. Immunoblotting
4.6. Confocal Microscopy
4.7. siRNA Treatment
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trefts, E.; Gannon, M.; Wasserman, D. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H. New perspectives on atherogenesis: Role of abnormal triglyceride-rich lipoprotein metabolism. Circulation 2002, 106, 2137–2142. [Google Scholar] [CrossRef] [PubMed]
- Sehayek, E.; Eisenberg, S. the role of native apolipoprotein B-containing lipoproteins in atherosclerosis: Cellular mechanisms. Curr. Opin. Lipidol. 1994, 5, 350–353. [Google Scholar] [CrossRef]
- Minehira, K.; Young, S.G.; Villanueva, C.J.; Yetukuri, L.; Oresic, M.; Hellerstein, M.K.; Farese, R.V.; Horton, J.D.; Preitner, F.; Thorens, B.; et al. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stpres or insulin sensitivity in mice. J. Lipid Res. 2008, 49, 2038–2044. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Acra, S. The role of lipid and lipoprotein metabolism in non-alcoholic fatty liver disease. Children 2017, 4, 46. [Google Scholar] [CrossRef]
- Barrows, B.; Parks, E. Contributions of different fatty acid sources to very low- density lipoprotein triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef]
- Donnelly, K.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Feingold, K. Introduction to Lipids and Lipoproteins. In EndoText; NBK305896; MDText.com, Inc.: South Dartmouth, MA, USA, 2021. [Google Scholar]
- Tiwari, S.; Siddiqi, S. Intracellular Trafficking and Secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef]
- Claude, A. Growth and differentiation of cytoplasmic membranes in the course of lipoprotein granule synthesis in the hepatic cell. I. elaboration of elements of the golgi complex. J. Cell Biol. 1970, 47, 745–766. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle in rat primary hepatocytes. Biochem. J. 2008, 413, 333–342. [Google Scholar] [CrossRef]
- Siddiqi, S.; Zhelyabovska, O.; Siddiqi, S. Reticulon 3 Regulates VLDL Secretion by Controlling VTV Biogenesis. Can. J. Physiol. Pharmacol. 2018, 96, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Siddiqi, S.; Zhelyabovska, O.; Siddiqi, S.A. Silencing of Small Valosin-containing Protein- interacting Protein (SVIP) Reduces Very Low Density Lipoprotein (VLDL) Secretion from Rat Hepatocytes by Disrupting its Endoplasmic Reticulum (ER)-to-Golgi Trafficking. J. Biol. Chem. 2015, 291, 12514–12526. [Google Scholar] [CrossRef]
- Tiwari, S.; Siddiqi, S.; Siddiqi, S. CideB Protein is Required for the Biogenesis of Very Low Density Lipoprotein (VLDL) Transport Vesicle. J. Biol. Chem. 2013, 288, 5157–5165. [Google Scholar] [CrossRef]
- Rahim, A.; Nafi-valencia, E.; Siddiqi, S.; Basha, R.; Runyon, C.C.; Siddiqi, S.A. Proteomic analysis of the very low density lipoprotein (VLDL) transport vesicles. J. Proteom. 2012, 75, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.P.; Gundry, R.L. The known unknowns of apolipoprotein glycosylation in health and disease. iScience 2022, 25, 105031. [Google Scholar] [CrossRef]
- Swift, L.L. Role of the Golgi apparatus in the phosphorylation of apolipoprotein B. J. Biol. Chem. 1996, 271, 31491–31495. [Google Scholar] [CrossRef]
- Tran, K.; Thorne-Tjomsland, G.; DeLong, C.J.; Cui, Z.; Shan, J.; Burton, L.; Jamieson, J.C.; Yao, Z. Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma McA-RH7777 cells. J. Biol. Chem. 2002, 277, 31187–31200. [Google Scholar] [CrossRef]
- Vukmirica, J.; Nishimaki-Mogami, T.; Tran, K.; Shan, J.; McLeod, R.S.; Yuan, J.; Yao, Z. The N-linked oligosaccharides at the amino terminus of human apoB are important for the assembly and secretion of VLDL. J. Lipid Res. 2002, 43, 1496–1507. [Google Scholar] [CrossRef]
- Alexander, C.; Hamilton, R.L.; Havel, R.J. Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J. Cell Biol. 1976, 69, 241–263. [Google Scholar] [CrossRef]
- Morré, D.; Ovtracht, L. Structure of rat liver golgi apparatus: Relationship to lipoprotein secretion. J. Ultrastruct. Res. 1981, 74, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Hossain, T.; Riad, A.; Parthasarathy, S.; Siddiqi, S.A. Mature VLDL triggers the biogenesis of a distinct vesicle from the trans-Golgi network for its export to the plasma membrane. Biochem. J. 2014, 459, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S. In vitro analysis of the very-low density lipoprotein export from the trans-Golgi network. Curr. Protoc. Cell Biol. 2015, 67, 11.21.1–11.21.17. [Google Scholar] [CrossRef] [PubMed]
- Bulow, V.; Gindner, S.; Baier, A.; Hehr, L.; Buss, N.; Russ, L.; Wrobel, S.; Wirth, V.; Tabatabai, K.; Quack, T.; et al. Metabolic reprogramming of hepatocytes by schistosoma mansoni eggs. JHEP Rep. 2022, 5, 100625. [Google Scholar] [CrossRef]
- Jiapeng, L.; Fike, K.R.; Dapper, C.; Klemba, M. Metabolism of host lysophosphatidylcholine in plasmodium falciparum-infected erythrocytes. BioRxiv 2023. [Google Scholar] [CrossRef]
- Shirey, C.; Ward, K.E.; Stahelin, R.V. Investigation of the biophysical properties of a fluorescently modified ceramide-1-phosphate. Chem. Phys. Lipids 2016, 200, 32–41. [Google Scholar] [CrossRef]
- Quinvivan, V.; Wilson, M.H.; Ruzicka, J.; Farber, S.A. An HPLC-CAD/fluorescence lipidomics platform using fluorescent fatty acids as metabolic tracers. J. Lipid Res. 2017, 58, 1008–1020. [Google Scholar] [CrossRef]
- Thibeaux, S.; Siddiqi, S.; Zhelyabovska, O.; Moinuddin, F.; Masternak, M.M.; Siddiqi, S.A. Cathepsin B regulates hepatic lipid metabolism by cleaving liver fatty acid binding protein. J. Biol. Chem. 2018, 293, 1910–1923. [Google Scholar] [CrossRef]
- Martin, G.; Danneberg, H.; Kumar, L.S.; Atshaves, B.P.; Erol, E.; Bader, M.; Schroeder, F.; Binas, B. Decreased liver fatty acid binding capacity and altered liver lipid distribution in mice lacking the liver fatty acid-binding protein gene. J. Biol. Chem. 2003, 278, 21429–21438. [Google Scholar] [CrossRef]
- Keler, T.; Sorof, S. Growth promotion of transfected hepatoma cells by liver fatty acid binding protein. J. Cell Physiol. 1993, 157, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.T.; Storch, J. Fatty acid transfer from liver and intestinal fatty acid-binding proteins to membranes occurs by different mechanisms. J. Biol. Chem. 1996, 271, 13317–13323. [Google Scholar] [CrossRef]
- Schachtrup, C.; Emmler, T.; Bleck, B.; Sandqvist, A.; Spener, F. Functional analysis of peroxisome-proliferator-responsive element motifs in genes of fatty acid- binding proteins. Biochem. J. 2004, 382 Pt 1, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Jamil, H.; Gordon, D.A.; Eustice, D.C.; Brooks, C.M.; Dickson, J.K., Jr.; Chen, Y.; Ricci, B.; Chu, C.H.; Harrity, T.W.; Ciosek, C.P.; et al. An inhibitor of the microsomal triglyceride transfer protein inhibits apoB secretion from HepG2 cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11991–11995. [Google Scholar] [CrossRef]
- Bakillah, A.; Nayak, N.; Saxena, U.; Medford, R.M.; Hussain, M.M. Decreased secretion of ApoB follows inhibition of ApoB-MTP binding by a novel antagonist. Biochemistry 2000, 39, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Rusiñol, A.; Verkade, H.; Vance, J. Assembly of rat hepatic very low density lipoproteins in the endoplasmic reticulum. J. Biol. Chem. 1993, 268, 3555–3562. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Gamble, M.V.; Conlon, D.; Liang, J.-S.; Ginsberg, H.N. The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoprotein in response to oleic acid occurs in the endoplasmic reticulum and not in the golgi in McA RH7777 cells. J. Biol. Chem. 2003, 278, 42643–42651. [Google Scholar] [CrossRef]
- Borchardt, R.; Davis, R. Intrahepatic assembly of very low density lipoproteins. Rate of transport out of the endoplasmic reticulum determines rate of secretion. J. Biol. Chem. 1987, 262, 16394–16402. [Google Scholar] [CrossRef]
- Kulinaki, A.; Rustaeus, S.; Vance, J.E. Microsomal triacylglycerol transfer protein is required for luminal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J. Biol. Chem. 2002, 277, 31516–31525. [Google Scholar] [CrossRef]
- Bamberger, M.; Lane, M. Possible role of the golgi apparatus in the assembly of very low density lipoprotein. Proc. Natl. Acad. Sci. USA 1988, 87, 2390–2394. [Google Scholar] [CrossRef]
- Higgins, J. Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the golgi complex. FEBS Lett. 1988, 37, 879–884. [Google Scholar] [CrossRef]
- Gusarova, V.; Seo, J.; Sullivan, M.L.; Watkins, S.C.; Brodsky, J.L.; Fisher, E.A. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J. Biol. Chem. 2007, 282, 19453–19462. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hao, J.; Zeng, J.; Sauter, E.R. Snapshot: FABP Functions. Cell 2020, 182, 1066. [Google Scholar] [CrossRef] [PubMed]
- Gajda, A.; Storch, J. Enterocyte Fatty Acid Binding Proteins (FABPs): Different Functions of Liver and Intestinal FABPs in the Intestines. Prostaglandins Leukot. Essent. Fatty Acids 2015, 93, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Smathers, R.; Petersen, D. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Siddiqi, S.A.; Siddiqi, S.; Mahan, J.; Lagakos, W.S.; Binas, B.; Gheyi, T.; Storch, J.; Mansbach, C.M., 2nd. Liver Fatty Acid- Binding Protein Initiates Budding of Pre-Chylomicron Transport Vesicles from Intestinal Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 17974–17984. [Google Scholar] [CrossRef]
- Mansbach, C.; Siddiqi, S. The Biogenesis of Chylomicrons. Annu. Rev. Physiol. 2010, 72, 315–333. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winterfeldt, K.; Tasin, F.R.; Siddiqi, S.A. Establishing the Role of Liver Fatty Acid-Binding Protein in Post-Golgi Very-Low-Density Lipoprotein Trafficking Using a Novel Fluorescence-Based Assay. Int. J. Mol. Sci. 2025, 26, 2399. https://doi.org/10.3390/ijms26062399
Winterfeldt K, Tasin FR, Siddiqi SA. Establishing the Role of Liver Fatty Acid-Binding Protein in Post-Golgi Very-Low-Density Lipoprotein Trafficking Using a Novel Fluorescence-Based Assay. International Journal of Molecular Sciences. 2025; 26(6):2399. https://doi.org/10.3390/ijms26062399
Chicago/Turabian StyleWinterfeldt, Kayli, Fahim Rejanur Tasin, and Shadab A. Siddiqi. 2025. "Establishing the Role of Liver Fatty Acid-Binding Protein in Post-Golgi Very-Low-Density Lipoprotein Trafficking Using a Novel Fluorescence-Based Assay" International Journal of Molecular Sciences 26, no. 6: 2399. https://doi.org/10.3390/ijms26062399
APA StyleWinterfeldt, K., Tasin, F. R., & Siddiqi, S. A. (2025). Establishing the Role of Liver Fatty Acid-Binding Protein in Post-Golgi Very-Low-Density Lipoprotein Trafficking Using a Novel Fluorescence-Based Assay. International Journal of Molecular Sciences, 26(6), 2399. https://doi.org/10.3390/ijms26062399