Abstract
Neovascularization is an important process in brain tumor development, invasion and metastasis. Several research studies have indicated that the VEGF signaling target has potential for reducing angiogenesis in brain tumors. However, targeting VEGF signaling has not met the expected efficacy, despite initial enthusiasm. This is partly because tumors cleverly use alternative growth factor pathways, other than VEGF signaling, to restore angiogenesis. Multi-target inhibitors have been developed to inhibit several receptor kinases that play a role in the development of angiogenesis. By simultaneously affecting various receptor kinases, these treatments can potentially obstruct various angiogenic pathways that are involved in brain cancer advancement, often offering a more holistic strategy than treatments focusing on just one kinase. Since 2009, the FDA has approved a number of multi-kinase inhibitors that target angiogenic growth factor receptors (e.g., VEGFR, PDGFR, FGFR, RET, c-KIT, MET, AXL and others) for treatment of malignant diseases, including brain cancer. Here, we present some recent results from the literature regarding the preclinical and clinical effects of these inhibitors on brain tumors.
1. Introduction
In brain tumors, a favorable microenvironment, characterized by hypoxia and extensive growth factor secretion, frequently induces a broad neovascularization, which in turn makes the tumor more forceful and resistant to treatment like radiation or chemotherapy. Angiogenesis is known as a hallmark of cancers; it has also been demonstrated that the glial tumors develop new capillary blood vessels capable of supporting the tumor’s growth [1]. This is a result of the imbalance between the pro-angiogenic and anti-angiogenic regulators [2]. Among the pro-angiogenic growth factors are vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF) and angiopoetin (Ang). Angiostatin and endostatin are the anti-angiogenic factors [3].
The receptors of the pro-angiogenic growth factors are vascular-endothelial growth factor receptors (VEGFRs), platelet-derived growth factor receptors (PDGFRs), fibroblast growth factor receptors (FGFRs) and Tie receptors. The binding of VEGF, PDGF and bFGF to the cognate receptors drives dimerization and stimulates the activation of several intracellular signaling pathways. For instance, VEGFR may induce the activation of the Ras/Raf/MEK/ERK pathway or the phospholipase-Cγ/protein kinase C(PLCy/PKC) pathway. These pathways are capable of regulating endothelial cell proliferation and migration, but also vascular permeability [4]. The phosphatidylinositol-3 kinase (PI3K)/phosphatase and tensin homologue (PTEN)/Akt/mammalian target of rapamycin (mTOR) is another important signaling pathway which is involved in vascular permeability, but also in endothelial cell survival. It may be activated by PDGFR, but also by PDGFR or bFGFR [5,6,7]. Tie-2 stimulates some signaling pathways through the Tie-2 receptor, also known as TEK, Ang-1. Some of these pathways are common with those activated by the dimerization of other pro-angiogenic growth factors such as Ras/Raf/MEK.ERK or PI3K/Akt/mTOR [8].
Brain tumor growth requests oxygen and nutrition, both supplied by new blood vessels. This process is triggered by hypoxia, which induces the expression of hypoxia-inducible factor-1 (Hif-1). Hif-1 is capable of activating the transcription of pro-angiogenic growth factors, including VEGF [9]. VEGF expression is correlated with cerebral microvascular proliferation: almost absent in low-grade gliomas and highly expressed in high-grade gliomas [10]. Instead, meningiomas, although highly vascularized, are less aggressive [11]. Both bFGF and PDGF are involved in recruiting peri-endothelial cells to vessels and are important pro-angiogenic regulators in brain tumors [12,13]. What is interesting is that physiological brain angiogenesis is regulated by a similar mechanism. The equilibrium between the pro-angiogenic and the anti-angiogenic factors plays a pivotal role in brain angiogenesis. During embryogenesis, VEGF expression is augmented in the neuroectoderm, while, in adult life, brain angiogenesis is absent and VEGF expression is at very low range. In highly malignant brain tumors, the VEGF level increases gradually to reach values similar to those initially found in embryogenesis [14,15,16].
Multi-target drug effectiveness in brain tumors is significantly influenced by the drug’s capacity to pass the brain–blood barrier (BBB). The BBB is mainly made up of endothelial cells, which form tight junctions that restrict anticancer drugs’ transport to the brain (Figure 1). Multi-target kinase inhibitors (i.e., axitinib, sorafenib, lenvatinib, pazopanib, sunitinib, cabozantinib, nintedanib and regorafenib) are relatively large, lipophilic molecules, and as a result, they have limited passive diffusion through the tight endothelial cell junctions that form the BBB. This limits their efficacy in treating brain tumors, particularly primary brain cancers or brain metastases. In malignant diseases, like brain cancer, several sites of the BBB may be disrupted by the loss of tight junction proteins, resulting in increased permeability (Figure 1A) [17]. The multi-kinase inhibitor axitinib was reported to assist BBB normalization/permeabilization in brain tumors, thus improving brain tumor response to treatment [18,19] (Figure 1A).
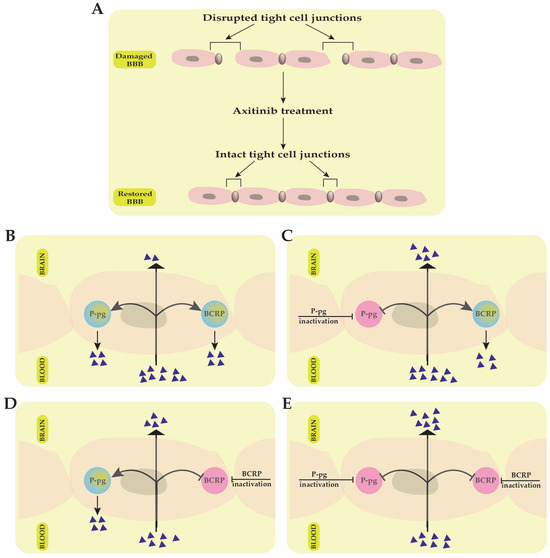
Figure 1.
The effectiveness of multi-target kinase inhibitors in treating brain tumors is significantly influenced by their ability to cross the blood–brain barrier (BBB). (A) In malignant brain tumors, the BBB structure can be disrupted due to the loss of tight junction proteins, increasing permeability. Treatment with axitinib helps normalize this barrier by restoring the integrity of tight cell junctions. (B) ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP), limit the penetration of multi-target kinase inhibitors into the brain by actively transporting them back into the bloodstream. (C) Inhibiting P-gp improves drug penetration into the brain, but BCRP continues to restrict drug transport into brain tissue. (D) Inhibiting BCRP increases intracerebral drug accumulation, but P-gp still reduces the efficiency of BBB crossing. (E) Simultaneous inhibition of both P-gp and BCRP allows improved penetration of multi-target kinase inhibitors, thereby enhancing the effectiveness of tumor treatment.
Interestingly, in a recent study by Kai Wang et al., published in 2024, axitinib was also found to have a protective effect on BBB alteration and cerebral ischemia-induced damage by attenuating the tight junction proteins’ injury [20].
The ATP-binding cassette efflux transporters further complicate BBB drug penetration in cancer. Two major proteins have also been shown to restrict drug transport across the BBB into the brain: ATP-binding cassette transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) (Figure 1). Several kinase inhibitors have been suggested to be substrates for these two multi-drug efflux transporters, further complicating their ability to traverse the BBB (Figure 1B) [21,22]. For example, in GB, the cooperation between the P-gp and BCRP transporters was demonstrated to restrict the ability of sunitinib to efficiently bypass the BBB, while inactivation of these efflux protein transporters improved the brain delivery and treatment efficacy of the drug (Figure 1C–E) [23].
Among the angiogenic inhibitors mentioned above, axitinib and sunitinib have been the best studied in terms of interaction with the BBB in cancer. Concerning brain tumors, several FDA-approved multi-target angiogenesis inhibitors (e.g., sorafenib, lenvatinib, pazopanib, cabozantinib, nintedanib, regorafenib) have undergone clinical trials and their ability to cross the BBB may be very different depending on the molecular weight, lipophilicity, bioavailability or systemic concentration of the drug. Unfortunately, there are no studies available so far that can clearly describe the BBB crossing mechanism for these angiogenic inhibitors.
Vasogenic oedema is another cause of morbidity in brain tumor patients. The same heterogenicity is encountered in medulloblastomas with different types of fenestration of the endothelial cells [24]. The consequence of this heterogenicity is the unequal distribution of drugs into brain tumors.
The hypothesis that angiogenesis was extremely important for the growth of brain tumors raised hopes for the therapeutic potential of anti-angiogenic therapies. Given the significance of the VEGF pathway in the development of cancer angiogenesis and the prevalence of VEGF in brain tumors, brain cancer therapies were nearly solely focused on blocking the VEGF pathway [25,26]. As a result, in May 2009, the Food and Drug Administration (FDA) approved the use of bevacizumab (Avastin, Genentech, Inc., South San Francisco, CA, USA) as a single agent for GBM patients with progressive disease following prior therapy [27]. Bevacizumab is a recombinant humanized monoclonal antibody, which targets all VEGF isoforms. The drug was tested either alone or in combination with other treatment modalities in GBM clinical trials. Although the single or combined regimen exceeded anterior records, the results were rather disappointing: the drug prolonged progression-free survival (PFS), and produced an improvement in neurological signs and a reduction in steroid intake, but did not improve overall survival (OS) [28,29,30,31]. In meningiomas, VEGF is also largely expressed, and the expression increases with meningioma grade. In this light, bevacizumab has shown some positive results. However, the prospective trials were rather small and there was a lack of control [32]. Other monoclonal antibodies that have been studied, either in preclinical or clinical studies, are tanibirumab or MSB0254. These drugs need further testing [33,34]. Our previous study showed that SU1498, a VEGFR inhibitor, had a cytotoxic effect on high-grade glioma cells (HGG), but the effect was rather limited [35]. Therapy resistance may be a cause of the limited response to treatment. For instance, there are patients diagnosed with GBM who do not respond to bevacizumab therapy because the tumor has intrinsic resistance to anti-angiogenic therapy. On the other hand, there are patients who have an initial response to Avastin treatment but may develop further resistance due to upregulation of other pro-angiogenic pathways, increased pericity coverage or increased invasiveness of tumor cells that determines the capacity to co-opt pre-existing brain blood vessels [36]. Some scientists have also discussed the role of biomarkers, which can be associated with tumor progression and angiogenesis but also with resistance to therapy [37,38,39,40].
Because angiogenesis is regulated by the crosstalk between multiple signaling molecules and various signaling pathways, dogma claims that multi-target drugs can potentially obstruct various angiogenic pathways. Single-target therapy generally targets proteins or genes with the purpose of stopping malignant cell survival and proliferation. Besides the positive effects, monotherapy is characterized by a lack of selectivity towards healthy cells. Another difficulty is the development of tumor cell resistance to single-target therapies [41]. On the other hand, the combination of drugs which have different mechanisms of action in the end, can lead to synergistic antitumor effects. This therapeutical possibility is also accompanied by negative effects like the summation of side effects of each drug, the high cost, or sometimes the side effects provoked by the interaction between drugs [42]. Multi-drug therapies have an important characteristic: one compound has high affinity to multiple targets at the same time. This is a way to reduce toxicity, the side effects, or costs, and, at the same time, to increase the efficacy of cancer treatment [43]. The purpose of anti-angiogenic therapy is the normalization of tumoral blood vessels, reduction or loss of hypoxia and the limitation of tumor invasion and metastasis, but also the improvement of the drug concentration in the tumor tissue [44]. For instance, bevacizumab, the first anti-angiogenic drug approved by the FDA, achieved some positive results either alone or in combination with other drugs or radiation therapy. However, the benefits of bevacizumab treatment did not meet expectations. Moreover, the FDA decided to withdraw bevacizumab for treatment of HER-2-negative breast cancer, in which the drug was not shown to be safe and efficient [45]. There are also other types of tumors where bevacizumab failed to demonstrate a significant anticancer effect: pancreatic cancer, gastric cancer and prostate cancer. One explanation could be the interference between the angiogenic signaling pathway with other signaling pathways [46].
This hypothesis led to the idea of developing multi-targeted anti-angiogenic therapies capable of overcoming resistance to anti-angiogenic monotherapy. Investigators embraced the idea of designing new, more potent and efficient anticancer drugs with multi-target properties [47]. Some of these treatments already have a success story. For instance, multi-target tyrosine kinases inhibitors like axitinib, sorafenib, lenvatinib, pazopanib, sunitinib, cabozantinib, nintedanib or regorafenib have already received FDA approval for the treatment of various solid tumors. Advanced stages of cancers like renal cell cancer, thyroid cancer, hepatocellular carcinoma, metastatic colorectal cancer, gastrointestinal stromal tumors and non-small cell lung cancer have seen improvements in survival due to this anti-angiogenic therapy. Similar results have been obtained by using multi-target monoclonal antibodies like aflibercept or ramucirumab for metastatic colorectal cancer, gastric cancer or non-small cell lung cancer [48]. Regarding brain tumors, these types of therapy are still under investigation.
Here, we focus on presenting the data regarding the efficacy of FDA-approved multi-target, anti-angiogenic drugs for brain tumor therapy. We will discuss preclinical in vitro and in vivo studies, but also the key clinical investigations.
2. Kinases as Targets for Anti-Angiogenic Therapy
VEGFR is among the tyrosine-kinases (TKRs) that are overexpressed in the endothelial cells of GBM. VEGF signaling is mediated through VEGFRs like VEGFR-1 (Flt-1), VEGFR-2 (KDR or FLK-1) or VEGFR-3 (FLT-4). These receptors have a transmembrane domain, an extracellular ligand-binding domain and a tyrosine kinase with an intracellular domain [49]. VEGFR-1 plays an important role in tumor angiogenesis and it is overexpressed in GBM cells [50], while in EGFRvIII-positive glioblastoma cells it has been observed that VEGFR-2 is mainly overexpressed [51]. It has also been demonstrated that, in GBM cells, a high level of VEGFR-3 is present [52]. The receptor, mainly VEGFR-2, is also overexpressed in meningiomas. In fact, VEGFR2 expression is significantly correlated with the WHO grade of the tumor [53].
Regarding the inhibition of angiogenesis in brain tumor patients since 2009, the US FDA has approved the inhibition of the VEGF-A/VEGFR-2 axis with bevacizumab for recurrent GBM. Since 2021, the drug has been included in European Association of Neuro-Oncology (EANO) guidelines due to its demonstrated improvement in quality of life and safety [54]. The drug has also been tested in meningioma patients but further clinical trials are needed [32].
The PDGF growth factors bind and activate two RTKs: PDGFR-α and PDGFR-β. This leads to receptor dimerization, transphosphorylation and activation of intracellular signaling pathways. The receptors have a transmembrane domain, a juxta-membrane domain, a kinase insertion domain, an intracellular domain and five extracellular Ig-like domains [55]. In normal conditions, they are important players involved in angiogenesis, growth and reproduction of endothelial cells, vascular maturation, revascularization and wound healing [56]. However, PDGFRs are expressed in a range of malignant tumor cells [57]. For instance, the amplification of PDGFR-α was found in GBM [58,59] and was associated with poor overall survival [60]. The overexpression of PDGFR-α in association with p53 mutation in GBM patients proved to be the least responsive to treatment [61]. A crosstalk between PDGF/PDGFR and other signaling pathways was also observed. An example of this is the PDGFRα- phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT) signaling pathway, which is activated in 70% of GBM [62]. The tumor invasion and angiogenesis promoted by AKT activation is a consequence of the overexpression of PDGFR-α induced by hypoxia [63].
PDGF-B and PDGFR-β are also overexpressed in GBM [64,65,66]. They are responsible for vessel maturation through the recruitment of peri-endothelial cells to vessels. PDGF-B stimulates glioma angiogenesis by inducing VEGF expression in tumor endothelial and perycite recruitment to growing vessels [67]. In fact, in astrocytoma grades I, II and III, angiogenesis is almost absent, while in GBM there is increased expression of PDGF-B and PDGFR-β. This event provokes the upregulation of VEGF in the tumor endothelium. In the end, all these processes determine the recruitment of pericytes [13]. Therefore, PDGF-B and PDGFR-β stimulate GBM angiogenesis. Meningiomas are also highly vascular tumors and most meningiomas express PDGFRβ [68].
The cognate receptors of FGFs are the four fibroblast growth factor receptors (FGFRs) 1 to 4. These receptors contain three extracellular immunoglobulin-like binding domains, a transmembrane domain and an intracellular domain which constitutes a two-part tyrosine kinase [69]. The binding of FGFs to FGFRs determines receptor dimerization and leads to autophosphorylation of tyrosine residues. The result is the activation of multiple signaling pathways like PI-3/AKT/mammalian target of rapamycin(mTOR) or the STAT3/NF-κB signaling pathway [70]. FGF2 is a growth factor involved in GBM vascularization [71]. The FGF2/FGFRs system is potentially an important target to block glioma angiogenesis [72]. FGF is also involved in meningioma angiogenesis [73].
The ANG family of growth factors has two receptor tyrosine kinases: Tie 1 and Tie2, also known as TEK [74]. The Tie 2 receptors are expressed by the endothelial cells. It has been observed that ANG1 is produced by pericytes and smooth muscle cells. This glycoprotein binds to the Tie 2 receptor and is very important in the process of recruitment of perycites and smooth muscle cells, but also in the remodeling of the vessel wall or for endothelial sprouting [75]. The other glycoprotein, ANG2, is produced by the endothelial cells. It also binds to Tie 2 receptor tyrosine kinase but does not activate the receptor. It is an antagonistic factor [76]. Unlike ANG1 and ANG2, ANGPTL4 does not bind to the ANG receptors Tie 1 and Tie 2 to mediate their biological functions. Its cognate receptors are unknown. However, it has been detected in a variety of organs or tissues like the liver, adipose tissues, kidneys, skin and intestines [77]. There are studies that emphasize the role of ANGPTL4 in tumor angiogenesis [78]. In 2010, Brunckhorst et al. demonstrated that ANGPTL4 promoted tumor angiogenesis in human GBM cells [79]. Also, ANG-1 and ANG-2 are expressed in GBM tumor cells and vessels [80]. The concomitant targeting of ANG-2 (with trebananib) and VEGF (with bevacizumab) improved vascular normalization and survival in GBM [81]. It has also been demonstrated that the ANG-2 plasma concentration of patients with meningiomas is rather high [82].
3. Evaluation of FDA-Approved Multi-Target Anti-Angiogenic Inhibitors in Preclinical Models of Brain Tumors
3.1. Axitinib
Axitinib, also known as AG013736 or Inlya (Pfizer Inc., New York, NY, USA) is a TK inhibitor [83]. This synthetic drug is an indazole derivative with a molecular weight of 386.47 Da (Figure 2) [84]. As a TK inhibitor, axitinib is capable of binding to the catalytic domain of VEGFRs. In fact, the drug is a potent inhibitor of VEGFR 1, 2 and 3 activities [85]. Regarding the mechanism of action, at picomolar concentration, it is capable of binding the intracellular ATP site of VEGFR in a competitive manner. The final result is the inhibition of signal transduction by VEGF. Axitinib also acts on the endothelial cells. Here, by blocking the VEGF/VEGFR pathways, it has the result of diminishing the phosphorylation of some protein kinases like AKT, or mitogen-activated protein kinases (ERK1/2). The phosphorylation of nitric oxide synthase (eNOS) is also affected. As a multi-target TK inhibitor, it can also suppress PDGFR and EGFR 1, 2 and 3, but also the gene cKIT. This is possible when the drug is administrated in picomolar concentrations [86] (Figure 3).
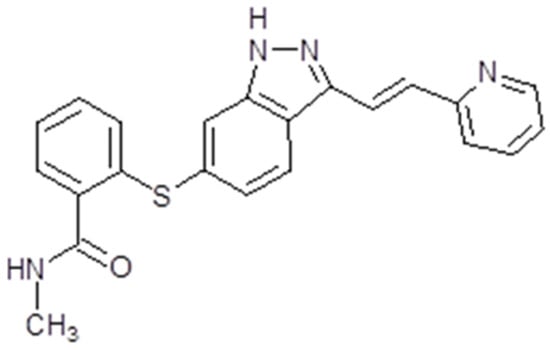
Figure 2.
Axitinib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
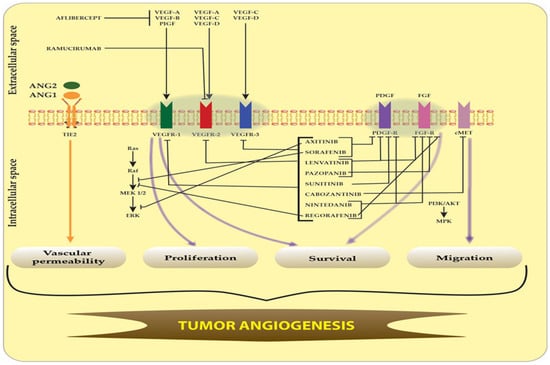
Figure 3.
Targeted inhibition pathways of VEGFR, PDGFR, FGFR, cMET and TIE-2 receptors by multi-kinase inhibitors. The blockade of VEGFR, PDGFR, FGFR, cMET and TIE-2 receptors and their signaling pathways by multi-kinase inhibitors leads to inhibition of glioma vascular permeability, migration, proliferation and survival within angiogenesis. The main FDA-approved multi-target angiogenesis drugs are axitinib, sorafenib, lenvatinib, pazopanib, sunitinib, cabozantinib, nintedanib, regorafenib, aflibercept and ramucirumab. Sharp arrows (→) illustrate stimulation while blunt arrows (┴) indicate inhibition.
The pharmacological presentation is tablets. After oral intake, the maximum plasma concentration occurs in about 4 h; therefore, the absorption process is rapid. Its metabolism is mainly in the liver, while the excretion is hepatobiliary [87]. Being metabolized in the liver, the pharmacokinetics are influenced by cytocrome P450 inducers like, for instance, phenytoin, or by CYP inhibitors like ketoconazole. It has a 58% oral bioavailability. Therefore, a dose reduction is needed in patients with hepatic diseases. The clearance of axitinib is not affected by the renal impairment [88].
Axitinib was tested in preclinical studies for different types of tumors: thyroid cancer [89], epithelial ovarian cancer [90], nasopharyngeal cancer [91], hepatocellular carcinoma (HCC) [92], non-small cell lung cancer [93], metastatic melanoma [94], pancreatic cancer, breast cancer [95] and colorectal cancer [96].
For the treatment of brain tumors, axitinib still had not been studied enough. In 2014, Wakimoto et al. demonstrated a direct cytotoxic effect against a number of patient-derived GBM stem cells and an endothelial cell line. The authors also proved that the drug has the capacity to prolong survival and decreased the tumor-associated vascularity in orthotopic GBM models [97]. In 2016, the capacity of axitinib to induce senescence-associated cell death and necrosis in glioma cell lines was demonstrated by Santoni et al. In the same study, the cytotoxic effect of the drug was demonstrated in combination with bortezomib mainly [98]. The drug is also capable of diminishing the proliferation and motility of human GBM cells [99]. In another in vitro study, axitinib retarded cell growth and had a cytotoxic effect on GBM cells [100]. In combination with oncolytic herpes simplex virus, the antitumor efficacy of the drug was enhanced in both immunodeficient and immunocompetent orthotopic GBM models [101]. In 2021, the combination of gemcitabine and axitinib was found to be cytotoxic for medulloblastoma cells. The same drug combination was well tolerated and tolerated in orthotopic human medulloblastoma xenograft mouse models [102]. One study in 2013 demonstrated the positive effects of axitinib in an organotypic in vivo model of human meningiomas [103] (Table 1).
3.2. Sorafenib
Sorafenib, also known as Nexavar® or BAY43-9006 (Bayer Pharmaceuticals Corp., West Haven, CT, USA and Onyx Pharmaceuticals Corp., Emeryville, CA, USA), is a TK inhibitor. It is a bi-aryl urea with a molecular weight of 464,825 g/mol (Figure 4). The drug was obtained through modification of the commercially available Raf-kinase inhibitor GK-00687. This small molecule is orally available and has multi-kinase inhibitory activity. This includes the capacity to inhibit pro-angiogenic RTKs like VEGFR2 [104]. Regarding its mechanism of action, the drug is capable of inhibiting RTKs like VEGFR, PDGFR or Raf in this way, impeding tumor growth and angiogenesis [105]. Also, it has been shown that sorafenib has the capacity to inhibit ERK signaling by reducing ERK phosphorylation. In nanomolar concentrations, it has an antiproliferative activity, while, in micromolar concentrations, it has antiproliferative activity [106]. The inhibition of proliferation was observed in human tumor cell lines that contained the mutations K-RAS or B-Raf [107] (Figure 3).
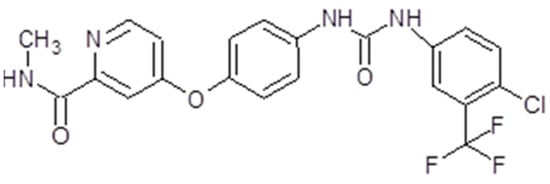
Figure 4.
Sorafenib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
The presentation of sorafenib is tablets for oral administration. Taken orally, the drug exhibits a bioavailability of 38–49%. The maximum plasma concentration is achieved in about 3 h. Sorafenib is metabolized primarily in the liver through oxidative metabolism and glucuronidation. Its metabolites are found in plasma, feces and urine. The metabolism of the drug is not altered in patients with chronic liver disease or if the creatinine clearance is under 30 mL/min [108].
Preclinical studies have demonstrated the capacity of sorafenib to inhibit tumor growth in many solid tumors [106]. The first studied effect of the drug was on renal cancer patients. Also, sorafenib was shown to reduce proliferation, and induced apoptosis in GBM both in vitro and in vivo. One study in 2006 demonstrated that the drug had the capacity to inhibit the proliferation of GBM cell lines, but also acted synergistically with bortezomib [109]. In 2010, the efficacy of the drug was tested both in vitro and in vivo. It was observed that sorafenib determined a dose-dependent inhibition of proliferation of GBM cells, also inducing autophagy and apoptosis, and inhibited the phosphorylation of signal transducer and activator of transcription 3 (Stat3), but also the expression of cyclins D and E. It was able to reduce angiogenesis, and was well tolerated [110]. In the same year, Yang et al. found out that sorafenib inhibited STAT3 signaling, contributing to growth arrest and inducing apoptosis in GBM cells [111]. In another in vitro study, Carra et al. also showed that sorafenib had an inhibitory effect on GBM cell proliferation, induced apoptosis by downregulating Mcl-1, and had a selective induction of cell death [112]. In 2016, the investigators detected no radio-sensitization and no chemo-sensitization of the drug in GBM cell lines. The drug had no effect on double treatment with irradiation and temozolomide [113] (Table 1).
Regarding meningioma cells, one in vitro study in 2014 demonstrated that sorafenib targets meningioma cell motility and brain invasion [114] (Table 1).
3.3. Lenvatinib
Lenvatinib, also known as Lenvima®, Kisplyx® or E7080 (Eisai Co., Ltd., Tokyo, Japan), is a multiple TK inhibitor. The drug is a quinoline derivative with a molecular weight of 426,853 g/mol, and is used as a mesylate salt (Figure 5). It is orally available. Lenvatinib has the capacity to inhibit multiple tyrosine kinases like VEGFR1-3, FGFR1-4, PDGFR, RET or KIT [115]. The drug proved to be an angiogenesis inhibitor and have effects on tumor cell migration and invasion, but does not influence tumor cell proliferation [116] (Figure 3).
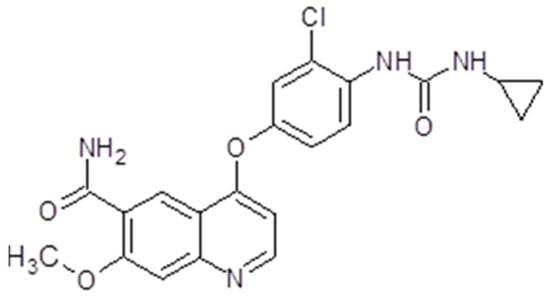
Figure 5.
Lenvatinib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
The presentation of lenvatinib is capsules for oral administration. It is absorbed rapidly and reaches maximum concentration in 1 to 4 h after ingestion. The bioavailability is 85–90%. After ingestion, the substance is bound to plasma proteins, albumin in particular, and is metabolized in the liver by the CYP3A4 enzyme. Mild-to-moderate liver impairment did not justify dose adjustment. In patients with severe liver damage, it is recommended to administrate 14 mg instead of 24 mg of lenvatinib [117]. The terminal half-life of the drug is 28 h. It is excreted via the feces (about two-thirds) and via urine (about a quarter) [118].
Several in vitro studies have demonstrated the anti-angiogenic effect of lenvatinib via inhibition of VEGFR2, VEGFR 3 and FGF1. Lenvatinib caused the regression of human small cell lung cancer cells through inhibition of angiogenesis [119]. It also inhibited VEGFR3-kinase in decreasing lymphatic vessel density within the metastatic lymph nodes after the resection of mammary breast tumor [115]. In one preclinical study in 2009, Ikuta et al. found that lenvatinib is capable of inhibiting the proliferation of endothelial cells and inhibited the progression of three cell lines of malignant pleural mesothelioma [120].
Lenvatinib has also been considered for brain tumor treatment. Regarding GBM, the first animal test was in 2017, when the drug was administrated to mice with advanced GB. In fact, the drug improved long-term survival [121] (Table 1).
3.4. Pazopanib
Another multiple TK inhibitor is pazopanib (GW786034, Votrient®, Glaxo SmithKleine, Brentford, United Kingdom).The drug is part of the group of indazolyl pyrimidines. For commercial use is available a hydrochloride salt insoluble at ph ≥ 4 and lightly soluble at ph = 1 [122]. The molecular weight is 437,517 g/mol (Figure 6).
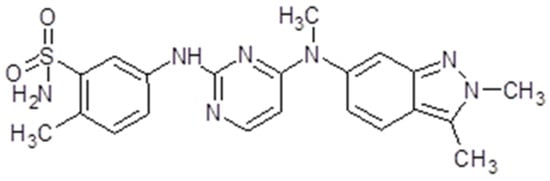
Figure 6.
Pazopanib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
The presentation of pazopanib is tablet or oral suspension. The recommended dose is 800 mg. At this dosage, the maximum plasma concentration is reached after 2–4 h [123]. The drug is bound to plasma proteins like albumin or α1 glycoprotein up to 99.99% [124]. The bioavailability is 21.4%. Pazopanib is metabolized mainly by oxidation via CYP3A4 and additionally by glucuronidation. In patients with mild hepatic impairment, the drug clearance is 50% lower; therefore, in this situation, the dosage should be diminished. Pazopanib should not be administrated in patients with severe hepatic impairment [125]. There are seven metabolites of the drug and the excretion is primarily through the feces (82.2%) [126].
Pazopanib is capable of inhibiting VEGFR 1 and 2, PDGFR α and PDGFRβ, c-KIT and FGR-1, 3 and 4 [127]. Through its capacity to inhibit VEGF, pazopanib inhibits the FGF-induced proliferation of human umbilical vein endothelial cell (HUVEC) cultures in vitro, but also impairs VEGF-induced angiogenesis as well as FGF-induced angiogenesis in mouse corneal micropocket [127]. Also, the drug induced the inhibition of tumor growth in xenograft models and the inhibition of VEGFR-2 phosphorylation [127] (Figure 3).
3.5. Sunitinib
Sunitinib (Sutent® or SU11248, Pfizer Inc, New York, NY, USA) is also known as INN-sunitinib malate. Is a member of the pyrroles and is a monocarboxylic acid amide. The molecular weight is 398.474 g/mol (Figure 7). The presentation of sunitinib is oral capsules, while the recommended dose is 50 mg. The time to maximum plasma concentration is 6 to 12 h [128]. Sunitinib binds to human plasma protein. The bioavailability is not influenced by food intake. The drug is primarily metabolized by cytochrome P450 3A4 to an active N-desethyl metabolite, which is also metabolized by the cytochrome P450 3A4. The drug is mainly eliminated through feces and only 16% is found in urine [128].
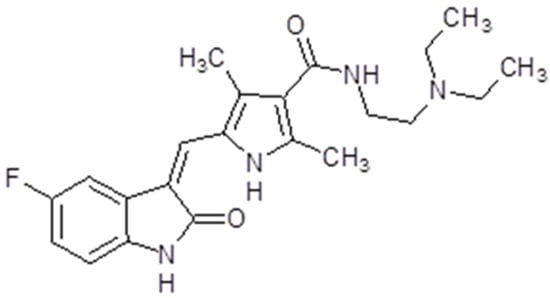
Figure 7.
Sunitinib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
Sunitinib is capable of inhibiting VEGFRs (VEGFR1, VEGFR2 and VEGFR3), PDGFRs (PDGFRα and PDGFRβ), Fms-like tyrosine kinase-3 receptor (FLT3), stem cell factor receptor (KIT) and the glial cell line-derived neurotrophic factor receptor (RET). This multi-target RTK inhibitor has an anti-angiogenic effect through VEGFR1, VEGFR2 and PDGFRβ-diminished signalization. It also has a potent antitumor activity on a variety of solid tumors. In preclinical studies, sunitinib demonstrated positive effects and tumor regression in various cancer cells, mainly gastrointestinal stromal tumors and advanced renal cell carcinoma [129] (Figure 3).
The anti-angiogenic effects of sunitinib were tested on orthotopic models of GBM in 2007. The drug had potent anti-angiogenic activity and prolonged survival [130]. In 2015, D’Amico et al. created a murine model of GBM expressing PDF-IRES-Cre retrovirus. The authors demonstrated that the combination of sunitinib and high-dose radiation was not only well tolerated but also delayed tumor growth. The association between sunitinib and low-dose radiation did not improve survival [131]. Another preclinical study was published in 2017. The authors demonstrated that the combination of CXCR4 antagonist and PRX177561 with sunitinib and bevacizumab inhibited tumor growth in preclinical models of human GBM [132]. In 2023, the results of a study that evaluated the activity of 16 new sunitinib derivatives in brain cancer cells and spheroids were published. The drug was demonstrated to have a significant impact on spheroid growth [133]. Ho et al. reported the results of a study on the combination of sunitinib and guanabenz in 2021. The effect of the angiogenenesis inhibitor on GBM cells was enhanced by guanabenz. Similar results were obtained on xenograft mice [134] (Table 1). For patients diagnosed with meningiomas, sunitinib had cytostatic and anti-migratory effects on human meningioma cells [135] (Table 1). In 2016, sunitinib was tested in a murine model with plexiform neurofibromatosis. The results were rather encouraging [136] (Table 1).
3.6. Cabozantinib
Cabozantinib (Cabometyx®, Cometriq®, Exelixis, Inc., Alameda, CA, USA) has a molecular mass of 501.514 g/mol (Figure 8). The presentation is oral capsules containing 20 mg, 40 mg or 60 mg of cabozantinib (S)-malate. The maximum plasma concentration is reached in 3 to 4 h, with a mean plasma concentration at a 60 mg dose [137]. Its clearance is rather variable. The drug is metabolized mainly by the P450 (CYP) 3A4 pathway but also by CYP2C9. The terminal half-life of the drug is 90–120 h. Therefore, hepatic impairment, medication or food intake may influence cabozantinib plasma concentrations [138].
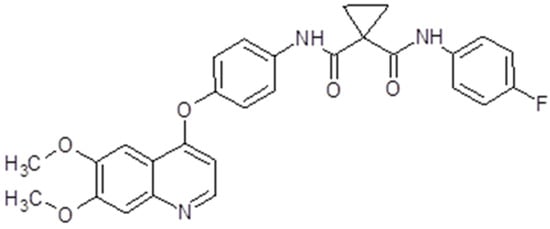
Figure 8.
Cabozantinib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
The drug proved to be a potent inhibitor of VEGFR2 and MET, but also Flt3, RET, KIT and AXL. The activation of multiple RTKs initiates downstream signaling pathways like PI3K/AKT, MAPK or JAK/STAT [139]. Unlike other anti-angiogenic multi-target TKIs, cabozantinib has a greater potency, making it a promising agent for investigation (Figure 3). As a result, it has been tested both in vitro and in vivo in solid tumors like medullary thyroid cancer [140], prostate cancer [141], osteosarcoma [142], schwannoma [143], gastrointestinal stromal tumor [144], pancreatic neuroendocrine tumors [145] and glioblastoma [146]. These in vitro and in vivo results led to clinical trials, which finally resulted in cabozantinib’s FDA approval for several solid tumors.
Cabozantinib (XL184) suppressed angiogenesis, cellular invasion and tumor growth in GBM cells [139]. The drug effects were also tested on c-MET-positive orthotopic E98 glioblastoma xenografts and it is a promising therapy for c-MET-positive glioma [146] (Table 1).
In meningiomas, cabozantinib inhibits the VEGFR2 and MET signaling pathways. In 2021, a clinical case was reported of regression of intracranial meningiomas after cabozantinib administration [147]. A phase II study of cabozantinib for patients with recurrent or progressive meningioma is now recruiting (Table 1).
3.7. Nintedanib
Nintedanib (Ofev®, Boehringer Ingelheim, Biberach, Germany) or Vargatef, also called BIBF1120, is a salt with ethanesulfonic acid. The molecular weight is 539.6248 g/mol (Figure 9). The presentation is 100 mg capsules for oral use. The bioavailability is 4.7%. The peak plasma concentration is reached in 2 to 4 h, while the inactivation is due to esterases. The drug is mostly excreted via bile and feces. Because it is a substrate for P-glycoprotein, several drugs influence its action either by diminishing or increasing it [148].
Figure 9.
Nintedanib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
As a multi-target tyrosine kinase inhibitor, nintedanib inhibits both PDGFR receptors along with VEGFR 1, 2, 3 and Flt3, but also FGFR 1, 2 and 3. Therefore, the drug initiates some downstream signaling pathways like PI3K/AKT, Ras/Raf/MEK/MAPK or FAK/Paxilin, inhibiting cell proliferation and angiogenesis [149] (Figure 3).
Nintedanib has been tested in vitro and in vivo on several solid tumors like lung adenocarcinoma [150], osteosarcoma [151], prostate adenocarcinoma [152], colorectal and hepatocellular carcinoma, as well as in gynecological tumors [153]. Although the drug has potential to be used in treatment of several tumors, its poor bioavailability is still a challenge. In 2025, Dang et al. investigated the effect of nintedanib on GBM cells and its mechanism of action. The inhibitor had a significant inhibitory effect on GBM cells and the drug delivery through the BBB was optimized through nanotechnology [154].
3.8. Regorafenib
Regorafenib (BAY-73 4506, Stivarga®, Bayer AG, Leverkusen, Germany) is a diphenylurea. The molecular weight is 482,82 g/mol (Figure 10). The presentation is 40 mg tablets for oral use with a bioavailability of 69–83%. The peak plasma concentration is reached in about 4 h after ingestion. Regorafenib is metabolized primarily in the liver. The result is two major and six minor metabolites. The two major metabolites M-2 and M-5 are pharmacologically active [155]. It is recommended that the drug is taken with a low-fat meal [156]. The unbound regorafenib or its metabolites are hydrolyzed in the enterohepatic circulation by the microbial actors in the gastrointestinal tract and then reabsorbed [157].
Figure 10.
Regorafenib chemical structure. The design was created with ACD/ChemSketch 2.0 (Freeware).
Regorafenib is a multi-target TKs inhibitor that targets receptor tyrosine kinases like VEGFR 1,2,3; TIE2; PDGFRβ; FGFR; KIT; REF and RAF. Therefore, the drug has anti-angiogenic, anti-metastatic and immunomodulatory effects [157] (Figure 3). It has been tested in vitro and in vivo on solid tumors like neuroblastoma [158], lung squamous cell carcinoma [159], osteosarcoma [160] and colorectal cancer [161].
Several preclinical studies have demonstrated the effect of regorafenib on glioma stem cells. In fact, the drug was able to determine a dose-dependent reduction of GSC’s pro-angiogenic ability [162,163]. Regorafenib’s dependent autophagy has also been reported [164], as well as apoptosis [165]. In some in vivo studies, regorafenib was able to inhibit tumor vascularization in GBM xenograft models. Also, the drug proved its anti-angiogenic and antitumor effects in GBM xenograft models [161,164]. A preclinical in vitro and in vivo study in 2017 established that regorafenib targets PDGFR and p44/42 ERK signaling. It also reduced cell motility and invasion, and mice with orthotopic meningioma xenografts presented a diminished tumor volume [166] (Table 1).
3.9. Aflibercept
Aflibercept (Eylea®, Zaltrap®, Regeneron Pharmaceuticals Inc., Westchester County, NY, USA) is a human monoclonal antibody. The molecular weight is 96.9 kilo Daltons (kDa). It is a dimeric glycoprotein. The VEGF-binding portion from the extracellular domains of human VEGF receptors 1 and 2 fused to the Fc portion of a human IgG1 immunoglobulin and formed a recombinant fusion protein [167], in this way acting as a VEGF-Trap. Aflibercept has a high affinity for VEGF A, B and PlGF (placental growth factor), and a strong bonding affinity to VEGFR. Also, it has the capacity to inhibit VEGF-A, VEGF-B, PIGF-1 and PIGF-2, as a consequence blocking their interaction with the cognate receptors and finally inhibiting angiogenesis [168] (Figure 3).
In cancer patients, the drug is administered 4 mg/kg intravenously every 2 weeks. It does not interfere with hepatic impairment, while renal dysfunction has no effect on Aflibercept clearance. The effects of the drug on various solid tumors have been tested in vitro and in vivo. Aflibercept’s effects have been investigated on tumors like retinoblastoma [169] and colorectal cancer [170].
One in vitro study demonstrated that VEGF-Trap treatment associated with radiation therapy significantly reduced tumor growth in a U87 subcutaneous xenograft model [171]. It was followed by an in vivo study in 2008 which reported that aflibercept therapy significantly prolonged the survival of glioma xenograft-bearing mice [172] (Table 1).
3.10. Ramucirumab
Ramucirumab (Cyramza®, Elli Lilly&Co, Indianapollis, IN, USA), also known as IMC-1121B, or LY3009806, is a human monoclonal antibody. The molecular weight is 143.6 kDa. The drug is a direct antagonist of VEGFR2. Therefore, by binding a specific epitope on the extracellular domain of VEGFR2, the drug blocks the binding of the receptor ligands (VEGF-A, VEGF-C and VEGF-D). In this way, the VEGF-stimulated phosphorylation of the receptor is prevented. Events like downstream ligand-induced proliferation, migration and permeability of human endothelial cells are no longer present. VEGF-mediated angiogenesis is inhibited [173] (Figure 3). The drug is administered intravenously 8 mg/kg up to 10 mg/kg depending on the regimen used. The clearance decreases as the dose of ramucirumab increases. The mean half-life at 8 mg/kg dose is 123 h for the first infusion and 318 h for the last infusion [174]. Renal deficiency or hepatic damage do not influence the drug dosage. Ramucirumab has in vitro and in vivo inhibitory effects on leukemia and ovarian cancer cell lines [154]. Also, in combination with paclitaxel, the monoclonal antibody was able to enhance the inhibitory effect of paclitaxel in gastric cancer cell lines [175]. No preclinical studies have been performed until now on brain tumors cells.

Table 1.
Main in vitro and in vivo studies that demonstrate the efficacy of anti-angiogenic multi-target therapies on brain tumors.
Table 1.
Main in vitro and in vivo studies that demonstrate the efficacy of anti-angiogenic multi-target therapies on brain tumors.
| Study | Study Area | Materials | Signaling Pathway | Molecular Mechanism |
|---|---|---|---|---|
| Lu et al., 2015 [97] | United States of America | Human glioma cells, Human GSCs, Human umbilical vein endothelial cells, Human brain microvascular endothelial cells, mice | No signaling pathway mentioned VEGFRs/PDGFR | Axitinib exhibits anti-angiogenic activity and prolongs survival of mice bearing orthotopic GBMs. |
| Morelli et al., 2016 [98] | Italy | Human glioma cells | No signaling pathway mentioned | Axitinib induces DNA damage response (DDR) characterized by γ-H2AX phosphorylation and Chk1 kinase activation leading to G2/M cell cycle arrest and mitotic catastrophe in glioma cell lines. Combined exposure to axitinib and bortezomib was more effective in inhibiting cell viability of all glioma cell lines. |
| Krcek et al., 2017 [99] | Germany | Human glioma cells | MAPKAP VEGFR2/phospholipase C/protein kinase C/ERK | The combination of axitinib and irradiation could be a potent strategy in the treatment of GBM. |
| Oprita et al., 2023 [100] | Romania | Human glioma cell line | No signaling pathway mentioned | Axitinib and sorafenib retarded GB1B cell growth in terms of dose and duration. |
| Saha et al., 2018 [101] | United States of America | GFP-positive mouse, Mouse brain microvascular endothelial cells (MBMECs), Human primary and recurrent GSCs | PDGFR/ERK/Akt | Axitinib has a dose-dependent anti-angiogenic effect while the antitumor effects of axtinib + G47Δ-mIL12 were mainly T-cell dependent. |
| Schwinn et al., 2021 [102] | Germany | Human medulloblastoma cells, mice | No signaling pathway mentioned | The combination of axitinib and gemcitabine has cytotoxic effects on medulloblastoma cells and favorable tolerability in xenograft models. |
| Yu et al., 2006 [109] | United America | Human glioma cells | Akt | Sorafenib interacts synergistically with bortezomib to induce apoptosis in glioma cells. |
| Siegelin et al., 2010 [110] | United States of America | Established or human glioma cells, mice | PI3K/AKT | Sorafenib has potent anti-glioma activity in vitro and in vivo. |
| Yang et al., 2010 [111] | United States of America | Human GBM cells | Akt, MAPK | The inhibition of STAT3 signaling by sorafenib contributes to growth arrest and induction of apoptosis in glioblastoma cells. |
| Carra et al., 2013 [112] | Italy | Human GBM cells | MAPK PI-3/Akt | Sorafenib reduces proliferation of glioblastoma cultures, and this effect depends, at least in part, on the inhibition of PI3K/Akt and MAPK pathways. Sorafenib significantly induces apoptosis/cell death via downregulation of the survival factor Mcl-1. Sorafenib has a selective action on glioblastoma stem cells. |
| Riedel et al., 2016 [113] | Germany | Human GBM cells | MAPK, Akt | Sorafenib had only minor effects on cell survival when administered alone and failed to enhance GBM cell killing by irradiation, TMZ or combined treatment, and instead rather caused resistance in some cell lines. |
| Wilisch-Neumann et al., 2014 [114] | Germany | Human meningioma cells | MAPK PI-3/Akt | Sorafenib reduce meningioma cell motility and brain invasion. |
| Jia Li et al., 2017 [121] | China | Human glioma cells, nude mice | No signaling pathway mentioned | Lenvatinib significantly increased apoptosis in glioma cell lines, and tumor growth was significantly inhibited in tumor-bearing mice. |
| Bouard et al., 2007 [130] | France | GBM cell lines, intra-cerebral xenograft models | No signaling pathway mentioned | Sunitinib had potent anti-angiogenic activity and prolonged survival. |
| D’Amico et al., 2012 [131] | USA | mice | No signaling pathway mentioned | The addition of sunitinib to radiotherapy enhances the effects of radiation in the brain and delays GBM growth without altering overall survival at the studied doses. |
| Gravina et al., 2017 [132] | Italy | GBM cells, subcutaneous xenografts, intracranial xenografts | No signaling pathway mentioned | An enhanced survival effect on GBM-bearing mice which were treated with a combination of PRX177561 and bevacizumab or sunitinib |
| Andrae et al., 2012 [135] | Germany | Human meningioma cells | PI3K/AKT ERK | Sunitinib strongly reduced meningioma cell migration in vitro, and had cytostatic effects. |
| Dang et al., 2025 [154] | China | GBM cells | No signaling pathway mentioned | Nintedanib exerted significant inhibitory effects on GBM cells. Drug delivery through nanotechnology may represent a new strategy for GBM treatment |
4. Clinical Trials Assessing the Effectiveness of FDA-Approved Anti-Angiogenic Inhibitors
4.1. Axitinib
The first clinical studies using axitinib were conducted on patients diagnosed with refractory solid tumors. The result of this phase I study determined the dose of 5 mg twice a day as the recommended dose for the next trials. The limited dosage was the result of toxicities like arterial hypertension [176]. It was followed by phase II and III clinical trials that tested the drug on patients diagnosed with kidney cancer [83,177,178]. As a result, in 2012, FDA approved axitinib for patients with advanced or metastatic clear cell carcinoma who had failed on one previous regimen therapy [86].
Following the promising preclinical results, a number of clinical trials studied the effect of axitinib on patients diagnosed with GBM. In 2019, Duernick et al. completed and published the results of a randomized phase II clinical trial which compared the effect of the drug as a single agent with the combination of axitinib and lomustin in patients diagnosed with recurrent GBM. Monotherapy with axitinib proved to be more efficient in improving the response rate and the PFS in recurrent GBM patients [179]. In 2020, Awada et al. published the results of another single-center phase II clinical trial (GliAvAx) with axitinib and avelumab for patients with recurrent GBM. Patients with prior treatment (surgery, radiation therapy, chemotherapy with temozolomide) were split into two groups in accordance with the dose of corticoids received. Those who had a daily dose of corticoid under 8 mg received the drug combination. Those who had a daily dose of corticoid of more than 8 mg, initiated therapy with axitinib and avelumab was added only if the corticoid therapy could be tapered under 8 mg. Although the combination of the two drugs had an acceptable toxicity, the clinical trial failed to meet its primary objective [180] (Table 2).
A phase II study of axitinib in patients with neurofibromatosis type 2 and progressive vestibular schwannomas showed that the drug had rather modest antitumor activity and greater toxicity when compared with bevacizumab [181].
Table 2.
Main clinical studies that used anti-angiogenic multi-targeted therapy for glioma treatment.
Table 2.
Main clinical studies that used anti-angiogenic multi-targeted therapy for glioma treatment.
| Trial Reference | Year | Who Tumor Grade, Histology Reference | Number of Patients | Clinical Trial Phase | Regimen | Signaling Pathways | Endpoints | Systemic Toxicity and Other Adverse Events |
|---|---|---|---|---|---|---|---|---|
| Duernik et al. [179] | 2018 | IV, rGBM | 79 | II | Axitinib 5 mg twice daily. Axitinib 5 mg twice daily and Lomustine 130 mg/m2 orally as a single dose every 6 weeks | not mentioned | 6 m PFS OS | neutropenia in the axitinib plus lomustine arm |
| Awada et al. [180] | 2020 | IV, rGBM | 54 | II | Axitinib 5 mg twice daily and avelumab 10 mg/kg intravenous every 2 weeks for patients with a daily dose of ≤8 mg of methylprednisolone. Patients with a higher dose of corticotherapy started with Axitinib 5 mg twice daily and added Avelumab 10 mg/kg intravenous every 2 weeks after 6 weeks if the corticotherapy dosage reached ≤8 mg. | not mentioned | 6 mPFS OS | dysphonia lymphopenia arterial hypertension diarrhea |
| Reardon et al. [182] | 2011 | IV, rGBM | 32 | II | Sorafenib 400 mg twice daily and temozolomide 50 mg/m | not mentioned | PFS-6 OS Toxicity of sorafenib and temozolomide The pharmacokinetics of sorafenib when combined with daily temozolomide | grade 2 and 3 elevation of amylase or lipase occurred in 2 and 4 patients grade 2 and 3 palmar-plantar erythrodysesthesia (PPE) occurred in 1(3%) and 6 (19%) patients fatigue rash infection electrolyte disturbances intracranial hemorrhage |
| Peereboom et al. [183] | 2013 | IV rGBM | 24 | II | Sorafenib 200 mg twice daily and tipifarnib 200 mg twice daily | Ras/Raf/Mek/ERK | Define DLT and determine the MTD | fatigue lipase diarrhea nausea pain rash AST ALT |
| Den et al. [184] | 2014 | WHO III grade or GBM | 15 | I | Radiation therapy consisted of a conventionally fractionated regimen to a total dose of 60 Gy, administered in 30 daily fractions of 2 Gy, with or without volumetric modulated arc therapy. Cohort 1 received a single daily oral dose of 200 mg, cohort 2 received 200 mg Sb BID and cohort 3 received 400 mg. After a break of 4 weeks, patients were treated in the maintenance phase with TMZ (150 mg m−2 on d1–5 for the first cycle of 28 days) followed for a total of up to six cycles of TMZ given at 200 mg m−2 on d1–5/28 if the first cycle was tolerated without significant side effects. Sorafenib restarted with 400 mg daily. | MAPK | The safety profile and tolerability of Sb when administered in conjunction with TMZ and RT and to establish the MTD of this combination. Secondary objectives were to evaluate pharmacokinetics (PKs), tumor response and survival. | thrombocytopenia fatigue hand–foot skin reaction |
| Nabors et al. [185] | 2011 | GBM, anaplastic astrocytoma, or anaplastic oligodendroglioma | 47 | I | Sorafenib 400 mg twice daily | Ras/Raf/MAPK | MTD | dermatological toxicity fatigue hyperglycemia hypertension hypophosphatemia nausea back pain joint pain. |
| Hottinger et al. [186] | 2014 | WHO grade III or GBM | 15 | I | Radiation therapy consisted of a conventionally fractionated regimen to a total dose of 60 Gy, administered in 30 daily fractions of 2 Gy, with or without volumetric modulated arc therapy. Three dose levels for Sb were planned as follows: cohort 1 received a single daily oral dose of 200 mg, cohort 2 received200 mg Sb and cohort 3 received 400 mg. After a break of 4 weeks, patients were treated in the maintenance phase with TMZ (150 mg m 2 on d1–5 for the first cycle of 28 days) followed for a total of up to six cycles of TMZ given at 200 mg m 2 on d1–5/28 if the first cycle was tolerated without significant side effects. Sorafenib was restarted on day 1 of the first cycle at 400 mg. | MAPK | The safety and maximum tolerated dose (MTD) of Sb in combination with radiation therapy (RT) and temozolomide (TMZ) | thrombocytopenia fatigue hand–foot skin reaction skin rush dyslipidemia diarrhea hypertension heart rate abnormalities |
| Chen et al. [187] | 2020 | GBM or gliosarcoma | 57 | I/II | Patients initially received sorafenib at 200 mg BID and erlotinib at 100 mg QD. | Ras/Raf/MAPK | MTD of sorafenib + erlotinib, charac terization of toxicities, and evaluation of drug interactions via pharmacokinetics studies. 6-month PFS (PFS6) | lymphocyte count decreased hypophosphatemia fatigue diarrhea lipase increased abdominal pain arthralgia dysphasia |
| Reardon et al. [188] | 2012 | rGBM | 32 | II | 24 mg Levantinib once daily in 28 cycles | Not mentioned | PFS-6 | hypertension fatigue headache proteinuria diarrhea fatigue hypertension one patient died due to pulmonary embolism |
| Lwin et al. [189] | 2020 | GBM | 31 | II | len 20 mg/d + pembro 200 mg Q3W for 35 cycles or until confirmed PD | Not mentioned | Efficacy and safety of lenvatinib plus pembro in pts with previously treated advanced solid tumors. Secondary endpoints included disease control rate (DCR), duration of response (DOR), PFS, and OS | manageable toxicity |
| Iwamoto et al. [190] | 2010 | rGBM | 35 | II | Pazopanib 800 mg orally daily on 28-day cycles | Not mentioned | PFS6 OS | anemia leukopenia lymphopenia neutropenia thrombocytopenia arterial hypertension fatigue excessive sweating weight loss decubitus ulcer dry skin flushing hand–foot syndrome hypopigmentation pruritus anorexia constipation diarrhea abdominal distension flatulence gastritis heartburn nausea CNS hemorrhage epistaxis elevated ALT elevated AST hyperbilirubinemia hypermagnesemia hypoalbuminemia hypophosphatemia joint or limb pain proteinuria abdominal pain thromboembolic event |
| Reardon et al. [191] | 2013 | rGBM | 75 | I/II | Pazopanib 400 mg q.d. plus lapatinib 1000 mg | Not mentioned | 6-month PFS Pharmacokinetics Maximum observed C Concentration time to maximum concentration Concentration 24 h post-dose | diarrhea fatigue hypertension nausea elevated ALT thrombocytopenia neutropenia elevated AST rash |
| Burzynski et al. [192] | 2014 | GBM | 11 | preliminary | Pazopanib 200 mg/daily–400 mg/daily Everolimus 5–10 mg/daily Sirolimus 1–3 mg daily Dasatinib 50 mg/daily Vorinostat 200–300 mg/daily Erlotinib 100–150 mg/daily Lapatinib 750 mg/daily Bevacizumab 2.5–10 mg/daily | Not mentioned | Further phase I/II clinical trials with PB in combination with pazopanib, dasatinib, everolimus and BVZ in patients with RGBM who failed standard surgery, radiation therapy and chemotherapy | anemia leukopenia thrombocytopenia hypertension fatigue sweating (diaphoresis) rash diarrhea dysphagia mucositis/stomatitis (clinical exam) hemorrhage, CNS alkaline phosphatase hyponatremia proteinuria neuropathy: sensory (paresthesia) pain: neck |
| Saada et al. [193] | 2024 | GBM | 35 | I/II | 800 mg orally daily on 28-day cycles | Not mentioned | PFS6 OS | hypertension increase ALT asthenia nausea diarrhea thrombopenia neutropenia anemia |
| Wuthrick et al. 2011 [194] | 2011 | Primary brain tumors and metastatic central nervous system malignancies | 15 | I | 37.5 mg sunitinib | Not mentioned | PFS OS | leukopenia thrombocytopenia anemia lymphopenia neutropenia hyponatremia hyperglycemia hypocalcemia hypercarbia hyperuremia hypokalemia elevated creatinine hyperbilirubinemia hypoproteinemia hypoalbuminemia elevated Alk. Phosphatase elevated ALT elevated AST fatigue pain dermatitis chest pain alopecia edema nausea anorexia diarrhea dyspepsia emesis mucositis seizure dysphasia drooling/difficulty chewing headache motor neuropathy DVT pulmonary embolism dyspnea epistaxis vaginal bleeding hypertension |
| Neyns, et al. [195] | 2011 | HGG | 21 | II | 37.5 mg sunitinib | Not mentioned | PFS OS | skin toxicity neutropenia thrombocytopenia lymphocytopenia |
| Duernick et al. [196] | 2015 | Anaplastic or low-grade glioma | 13 | II | Sunitinib malate (Sutent, Pfizer) was administered at a daily dose of 25 mg for 28 consecutive days followed by a 14-day treatment-free interval. Lomustine (CCNU) was administered as a single dose (80 mg/m2) on day 14 of the 6-week treatment cycle | Not mentioned | PFS OS | fatigue mucositis thrombocytopenia lymphopenia neutropenia |
| Wetmore et al. [197] | 2016 | Recurrent or refractory high-grade glioma or ependymoma | 30 | II | Sunitinib, 15 mg/m2 | Not mentioned | PFS OS | alanine aminotransferase increased aspartate aminotransferase increased lipase increased lymphocyte count decreased neutrophil count decreased serum amylase increased white blood cell decreased diarrhea fatigue intracranial hemorrhage rash maculo-papular skin and subcutaneous tissue disorders—Other (rash, acne) |
| Wuthrick et al. [198] | 2014 | Recurrent high-grade glioma | 11 | I | 37.5 mg sunitinib The fSRT doses delivered ranged from 30 to 42 Gy in 2.5- to 3.75-Gy fractions | (PI3K)-Akt-mTOR | Safety and toxicity profile of continuous daily-dosed sunitinib when combined with hypofractionated stereotactic RT (fSRT) for recurrent high-grade gliomas (rHGG). | leukopenia anemia thrombocytopenia fatigue candidiasis nausea vomiting diarrhea hypocalcemia hyponatremia acid reflux xerostomia hypoproteinemia hypochloremia elevated ALT elevated AST pain cough alopecia aphasia stomatitis anorexia hyperglycemia elevated ALP muscle weakness esophagitis hypertension |
| Faye et al. [199] | 2023 | MGMT promoter GBM | 37 | II | 12.5 mg of daily sunitinib for 7 days, followed by concurrent chemoradiation plus 12.5 mg sunitinib, then adjuvant TMZ | Not mentioned | PFS OS safety | fatigue anemia leukopenia lymphocytopenia neutropenia thrombocytopenia pulmonary embolism deep vein thrombosis appetite loss (anorexia) constipation diarrhea dysgeusia (taste alteration) increased liver enzyme increased creatinine hyperglycemia nausea vomiting (emesis) weight loss (anorexia) seizures speech impairment ataxia muscle atrophy/weakness neuropathy cognitive disturbance confusion mood (depression/anxiety) dizziness drowsiness headache fever brain infection alopecia hypertension tachycardia coughing dyspnea shortness of breath on exertion |
| Janssen et al. [200] | 2024 | Recurrent GBM | 55 | II/III | High-dose intermittent sunitinib 300 mg once weekly (Q1W, part 1) or 700 mg once every two weeks (Q2W, part 2) or lomustine | Not mentioned | PFS OS | blurred vision diarrhea dysgeusia fatigue flu-like symptoms headache hypertension hypothyroidism mucositis oral muscle weakness lower limb musculoskeletal disorders nausea oral pain palmar–plantar erythrodysesthesia syndrome rash septic bursitis skin discoloration syncope tooth infection vertigo vomiting anemia lymphocyte count decreased neutrophil count decreased platelet count decreased white blood cell decreased alanine aminotransferase increased alkaline phosphatase increased aspartate aminotransferase increased GGT increased |
| Kaley et al. [201] | 2014 | Anaplastic meningioma | 36 | II | Sunitinib was administered at 50 mg/d for days 1–28 of every 42-day cycle | Not mentioned | PF6 Radiographic response safety | CNS hemorrhage thrombotic microangiopathy neutropenia hypophosphatemia fatigue thrombocytopenia lymphopenia leukopenia hypertension headache ALT AST dehydration pain, abdomen hyperglycemia rash, hand-foot reaction vomiting pancreatitis hypocalcemia confusion diarrhea creatinine hypomagnesemia prolonged QTc interval right ventricular enlargement thrombosis/embolism hyperuricemia gastrointestinal perforation |
| Cardona et al. [202] | 2019 | WHO II or WHO III meningiomas | 31 | II | Octreotide acetate LAR [O]/everolimus [E] (30 mg IM q28 days/10 mg PO q/day), sunitinib [Su] (50 mg PO q/day for days 1–28 of 42 days) or bevacizumab [Bev] (10 mg/kg IV days 1 and 15) | Not mentioned | PFS OS | fatigue hypothyroidism |
| Schiff et al. [203] | 2016 | HGG | 26 | I | Cabozantinib at a dose of 40 mg or 60 mg daily | MET | Grade 3/4 adverse events Maximum tolerated dose | thrombocytopenia fatigue constipation nausea diarrhea elevated ALT neutropenia elevated AST leukopenia elevated LDH hypertension |
| Cloughesy et al. [204] | 2018 | Progressive GBM | 152 | II | Cabozantinib starting dose of 140 mg/day, but the starting dose was amended to 100 mg/day because of toxicity. | MET | PF6 OS | fatigue diarrhea decreased appetite PPES nausea headache constipation hypertension weight decreased dysphonia AST increased ALT increased convulsion LDH increased hypophosphatemia confusional state stomatitis vomiting abdominal pain thrombocytopenia pain in extremity insomnia gait disturbance hair color changes leukopenia lipase increased cough dysgeusia anxiety oral pain depression dry skin hemiparesis dyspepsia edema peripheral oropharyngeal pain rash hypokalemia neutropenia dyspnea dizziness cognitive disorder lymphopenia proteinuria aphasia vision blurred bilirubin increased epistaxis skin discoloration |
| Muhic et al. [205] | 2013 | Recurrent GBM | 13 | II | Nintedanib was given orally at a dose of 200 mg twice daily | Not mentioned | PF OS | fatigue loss of appetite, diarrhea nausea |
| Norden et al. [206] | 2015 | HGG | 36 | II | Nintedanib was given orally at a dose of 200 mg twice daily | Not mentioned | PF OS | treatment was well tolerated |
| Lombardi et al. [207] | 2019 | Relapsed GB | 124 | II | Regorafenib 160 mg once daily for the first 3 weeks of each 4-week cycle or lomustine 110 mg/m2 once every 6 weeks | Not mentioned | PFS OS toxicity | hand–foot skin reaction increased lipase blood bilirubin increased |
| Chiesa et al. [208] | 2022 | Recurrent GBM | 30 | II | Regorafenib was administered orally at a dose of 160 mg/day for the first 3 weeks of each 4-week cycle | MAPK pathway | PFS OS | thrombocytopenia fatigue hand–foot syndrome diarrhea hyperbilirubinemia |
| Rudà et al. [209] | 2022 | Recurrent GBM | 66 | I/II | Regorafenib daily dose was gradually increased from 80 to 160 mg across the first 2 cycles. | Not mentioned | PFS OS toxicity | grade 3–4 toxicity |
| Fasano et al. [210] | 2023 | Recurrent GBM | 56 | II | 160 mg of regorafenib (four 40 mg tablets) per day orally for three weeks in a four-week cycle | Not mentioned | PFS OS | hand–foot skin reaction rash/desquamation piastrinopenia neutropenia hypertension fatigue voice changes vomiting hepatic AEs, aspartate aminotransferase elevation hyperbilirubinemia proteinuria fever cardiac diarrhea |
| Nayak et al. [211] | 2011 | HGG | 59 | I | Aflibercept 4 mg/kg every 2 weeks | VEGF PIGF | Maximum tolerated dose Toxicities | abdominal pain Alanine aminotransferase increase Aspartate aminotransferase increase alkaline phosphatase increase bilirubin increase Gamma glutamyl transferase increase arthralgia colonic perforation colitis dehydration fatigue headache hypertension hypokalemia hyponatremia seizure nausea lung infection peripheral nerve infection 1 urinary tract infection vascular access complication leukopenia lymphopenia neutropenia thrombocytopenia |
| De Groot et al. [212] | 2011 | Malignant glioma | 58 | II | Aflibercept 4 mg/kg was administered intravenously on day 1 of every 2-week cycle | VEGF PIGF | PF6 Overall radiographic response | ataxia CNS ischemia confusion dysphagia fatigue GI hemorrhage hand–foot syndrome headache hypertension hyponatremia hypophosphatemia hypoxia increased LFTs lymphopenia mucositis neutropenia pain pericarditis proteinuria rash thrombosis/embolism wound complication |
4.2. Sorafenib
Sorafenib treatment showed promising results in the phase I [213], phase II [214] and III [215] clinical trials, which led in 2005 to FDA approval of sorafenib for advanced renal cancer treatment. After that, in 2007, sorafenib was approved by the FDA as first-line treatment for patients with advanced HCC [216], while in 2013 the FDA approved sorafenib for the treatment of radioactive iodine-resistant (RAI-R) metastatic well-differentiated thyroid cancer (DTC) [217].
The drug was tested in clinical trials for some other solid tumors like desmoid tumors [218], pancreatic cancer [219], on behalf of the Italian Group for the Study of Digestive Tract Cancer (GISCAD), melanoma [220], non-small cell lung cancer (NSCLC) [221] and breast cancer [222].
A phase I clinical trial in 2011 determined the maximum tolerated dose of sorafenib in patients diagnosed with brain tumors (GBM, anaplastic astrocytoma or anaplastic oligodendroglioma), progressive or recurrent, after radiation therapy with or without chemotherapy. The maximum tolerated dose (MTD) of sorafenib given orally on a continuous basis was established as 600 mg in patients with malignant glioma who were concurrently receiving enzyme-inducing antiseizure drugs and 800 mg in those who were not [185]. However, when tested in clinical trials either alone or in combination with temozolomid or erlotinib, in patients with recurrent or progressive GBM, the effect of sorafenib was rather disappointing [182,183]. Regarding the association between sorafenib and X-irradiation there are some promising results [186]. A phase I/II clinical trial determined the MTD and 6-month PFS of patients diagnosed with GBM or gliosarcoma and treated with sorafenib and erlotinib. The results were rather disappointing [187] (Table 2). In 2014, a phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas also had disappointing results [223].
4.3. Lenvatinib
In clinical studies, lenvatinib demonstrated its efficiency for the treatment of several solid tumors. In one phase II clinical trial in 2015, Lenvatinib had a positive effect in patients with radiation-refractory differentiated thyroid cancer. The patients had a PFS of 12.6 months [224]. Also in 2015, a phase III clinical trial with lenvatinib was conducted in patients with radiation-refractory thyroid cancer with radiographic progression within the prior 12 months. In this situation, the median PFS was 18.3 months, and the toxicity was significant [225]. As a result, in 2015, the FDA approved lenvatinib for the treatment of radiation-refractory thyroid cancer [226]. The same drug in combination with everolimus was approved by the FDA in 2016 for the treatment of clear-cell renal carcinoma [227]. In 2018, lenvatinib received approval as a first-line treatment for patients with unresectable HCC [228]. In September 2019, the FDA approved the combination of pembrolizumab and lenvatinib for patients with certain types of endometrial carcinoma [229]. For several other solid tumors like melanoma [230], lung adenocarcinoma [231], ovarian cancer [232], breast cancer [233], biliary tract cancer [234] and pancreatic cancer [235] the results were also promising.
The first clinical trial that studied the effect of lenvatinib treatment on patients with progressive GBM on bevacizumab was in 2012. The results were rather modest: the PFS rate was 1.9 months, while the median overall survival (OS) was 4.11 months. Also, the tumor volume significantly decreased one day after the drug administration, which was considered to be a consequence of of lenvatinib’s capacity to decrease vascular permeability and tumor vascularity [188]. In 2020, the results of a phase II study of lenvatinib plus pembrolizumab were published. The association was demonstrated to have promising antitumor activity, and reduced toxicity. The median duration of response was 3.2 [189] (Table 2).
4.4. Pazopanib
The first FDA approval for pazopanib was in 2009 for the treatment of advanced renal cancer. This was a result of phase III clinical trials [236]. After that, the drug was approved by the FDA in 2012 for the treatment of soft tissue sarcoma [237].
Regarding GBM, a phase II trial of pazopanib did not prolong PFS in patients with recurrent GBM [190]. In 2013, Reardon et al. published the results of a phase I/II trial of pazopanib in association with lapatinib for patients with relapsed GBM. However, the results were rather disappointing, and the antitumor activity of this combination of drugs was limited [191]. In 2014, Burzynski et al. published another study of pazopanib in combination with phenylbutyrate in patients with GBM. The combination was tolerated. The conclusion was that further studies were needed [192].
The results of a phase I/II study of pazopanib and temozolomide in patients newly diagnosed with GBM were reported in 2024. This was the PAZOGLIO trial, of which the objective was to evaluate the safety of this drug combination. In fact, the patients were treated after the partial or complete resection of the tumor. The administration of the drug combination was carried out during the maintenance phase, as the Stupp regimen defines it. Saada et al. concluded that, after the evaluation of safety, which represents the first phase of the study, the association between temozolomide and pazopanib in a dose of 600 mg daily is feasible. However, phase II of the study, regarding the efficacy of the combination, is now enrolling patients [193] (Table 2).
4.5. Sunitinib
The first FDA approval for sunitinib was in 2006 for gastrointestinal stromal tumors and advanced renal cell carcinoma [238,239]. It was followed by the FDA approval of the drug for rare types of pancreatic cancer, which was a result of a phase III clinical trial [240,241].
A phase I clinical trial studied the effect of the combination of sunitinib and radiotherapy in patients with primary brain tumors. The drug had acceptable toxicity and the adverse events were limited. This study encouraged the researchers to progress to phase II clinical trials [194]. In 2011, Neyns et al. published the results of a phase II study of sunitinib malate in patients with recurrent high-grade glioma. The drug had insufficient activity [195]. Since then, there have been many clinical trials that have reported similar results. The drug was tested either as a single agent [242,243,244,245] or in combination with irinotecan [246]. Regarding the combination between sunitinib and lomustine, in 2015, Duernick et al. published the results of a clinical trial including 13 patients treated with this combination of drugs. Although this regimen was tolerated, it was not active enough [196]. Another phase II clinical trial in 2016 evaluated treatment with sunitinib for children diagnosed with recurrent or refractory high-grade glioma or ependymoma. The drug was sufficiently tolerated but did not have enough efficacy [197]. In 2014, a pilot study followed the effect of the combination of radiation therapy and sunitinib in previously irradiated recurrent high-grade glioma patients. The results were rather encouraging, with acceptable 6-month PFS [198]. In 2023, Faye et al. concluded after a phase II clinical trial that the concurrent administration of sunitinib, temozolomide and radiotherapy in newly diagnosed patients with MGMT unmethylated GBM might be beneficial [199]. In 2024, the STELLAR phase II/III clinical trial analyzed the intermittent administration of high-dose sunitinib in patients diagnosed with recurrent GBM. However, the conclusion was that this regimen failed to improve the outcome for the patients [200] (Table 2).
A phase II clinical trial of sunitinib for patients diagnosed with recurrent and progressive atypical and anaplastic meningioma demonstrated in 2014 that the drug is active in meningioma patients [201]. In 2019, a comparative survival and molecular marker analysis evaluated the efficacy of sunitinib and octreotide/everolimus on patients with malignant meningiomas. The results were similar [202] (Table 2).
4.6. Cabozantinib
The first FDA approval of cabozantinib was in 2017 for previously untreated advanced renal cancer carcinoma [247]. It was followed in 2019 by FDA approval of the drug for previously treated hepatocellular carcinoma [248]. In 2021, the association between nivolumab and cabozantinib was FDA approved as a first-line treatment for patients with advanced renal cancer carcinoma [249]. Also in 2021, the drug was approved for previously treated radioactive iodine-differentiated thyroid cancer [250].
A phase I trial of cabozantinib plus temozolomide and radiotherapy in patients newly diagnosed with high-grade glioma and pre-treated with radiation therapy established the dose of 40 mg daily. The drug was tolerated [203]. In 2018, a phase II clinical study of cabozantinib in patients with progressive GBM demonstrated a modest clinical activity [204] (Table 2).
A phase II study of cabozantinib for patients with recurrent or progressive meningioma is now recruiting (Table 1).
4.7. Nintedanib
The results of preclinical studies led to clinical trials for several solid tumors. Until now, the drug has not been FDA approved for any solid tumor. However, nintedanib received its first FDA approval in 2014 for the treatment of idiopathic pulmonary fibrosis [251]. In 2019, the drug received FDA approval for the treatment of interstitial lung disease associated with systemic sclerosis or scleroderma [252]. This was followed in 2020 by the FDA approval of nintedanib for treatment of chronic fibrosing interstitial lung disease with progressive phenotype [253].
Regarding GBM, one phase II open-label study determined that the single-agent nintedanib, at a dose of 200 mg twice a day, had a limited and clinically non-relevant antitumor activity [205]. Another phase II clinical trial in 2015 found that treatment with nintedanib, although tolerated, was not active against high-grade glioma, either prior to or after bevacizumab therapy [206] (Table 2).
4.8. Regorafenib
The result of preclinical studies was the investigation of regorafenib’s effect in a series of clinical trials, which resulted in the FDA’s approval of the drug for treatment of various tumors. The first FDA approval for regorafenib was in 2012 for patients with metastatic colorectal cancer [254]. The same year, the drug was approved for patients with advanced colorectal cancer. Next, the drug received approval to be used for advanced gastrointestinal stromal tumors in 2013 [238]. Since 2017, regorafenib has received approval to be used for hepatocellular carcinoma patients [255].
REGOMA is a clinical trial of which the aim was to compare regorafenib with lomustin for GBM patients. The drug significantly improved OS [207]. Similar results were obtained in 2022 by Chiesa et al. [208]. In another study, Ruda et al. reported the efficacy of the drug with improvement in PFS and OS [209]. Regorafenib had similar efficiency in elderly GBM patients [210]. A couple of studies followed the MRI evolution of GBM in patients treated with regorafenib. The conclusion was to assess the therapy with caution [256]. The drug was also evaluated in a phase I clinical trial, in combination with temozolomide with or without radiation therapy in patients with newly diagnosed MGMT-methylated, IDH-wild=type GBM [257]. Nowadays, according to with ClinicalTrials.gov, there are several ongoing clinical trials with regorafenib for recurrent GBM patients. For instance, the global, innovative learning environment (AGILE) is trying to evaluate multiple therapies for newly diagnosed or recurrent GBM patients [258] (Table 2).
The MIRAGE trial studies regarding the effect of regorafenib for recurrent grade 2 and 3 meningiomas are currently recruiting patients.
4.9. Aflibercept
The results of preclinical studies caused investigators to evaluate the responses of patients to aflibercept. Therefore, they tested the drug in clinical trials, and this led to the FDA approval of aflibercept for cancer patients. In the end, the drug was FDA approved for metastatic colon cancer in 2012 [259,260].
The first results of a phase I clinical trial of aflibercept and temozolomide in patients newly diagnosed with high-grade gliomas were published in 2011. This three-arms study evaluated the drug’s safety along with the maximum tolerated dose (MTD). The patients enrolled in arms 2 and 3 of the study were diagnosed with anaplastic glioma, including anaplastic astrocytoma, anaplastic oligodendroglioma, anaplastic mixed oligoastrocytoma or malignant astrocytoma, while in arm 1 only patients newly diagnosed with GBM were admitted. The combination was tolerated. As a result, the investigators recommended a phase II clinical trial of aflibercept with radiotherapy and concomitant temozolomide [211,261]. In the same year, the results of this phase II clinical trial were reported. As monotherapy, aflibercept had moderate toxicity and minimal benefits regarding OS in patients with recurrent GBM [212] (Table 2).
4.10. Ramucirumab
Ramucirumab proved to have a potential therapeutic role in solid tumors either as a single agent or in association with other chemotherapeutics [262]. As a new therapeutic option in solid tumors, the drug was tested in a series of phase I clinical trials evaluating the maximum tolerated dose as well as the toxicity [174]. All these positive results were followed by phase II and III clinical trials. Finally, in 2014, the FDA approved ramucirumab in combination with paclitaxel for advanced gastric cancer after prior chemotherapy. The RAINBOW, a double-blind randomized phase III trial, concluded that ramucirumab in combination with paclitaxel significantly prolonged OS in patients with advanced gastric cancer [263]. In the same year, the FDA expanded its approval to include treatment of aggressive non-small cell lung cancer [264]. Since then, the drug has received approval for metastatic colon cancer (as a second-line treatment in association with FOLFIRI) in 2015 [265], hepatocellular carcinoma in 2019 [266] and as a first-line treatment for metastatic EGFR-mutated non-small cell lung cancer in 2020 [267].
The preliminary results of a phase II clinical trial reported in 2013 demonstrated that the drug had a potent anti-angiogenic effect in patients with recurrent GBM [268]. This was a phase II trial which studied ramucirumab or IMC-3G3 (an anti-PDGFRα monoclonal antibody) as anti-angiogenic therapy in patients with recurrent GBM. The primary objective of the study was to assess the PFS rate at 6 months after treatment. The secondary objectives were to determine the acute and late toxicities, the tumor response rate, OS and to describe the pharmacokinetic and pharmacodynamic profiles. The patients were enrolled in two groups. Group 1 comprised recurrent GBM patients, 40 of whom received ramucirumab. The PFS at 6 months was 12.5 superior to those receiving IMC-3G3, for whom it was 7.5. The OS in the ramucirumab arm was 49.5. The data are available on ClinicalTrials.gov, ID NCT00895180: Ramucirumab or Anti-PDGFR Alpha Monoclonal Antibody IMC-3G3 in Treating Patients with Recurrent Glioblastoma Multiforme.
5. Conclusions
In preclinical studies and clinical trials, multi-kinase angiogenic inhibitor therapy has been established as a potential treatment for several types of cancer. Although the efficacy of multi-target angiogenic therapy has been confirmed in vitro and in vivo, it still faces several challenges concerning drug-induced adverse reactions due to toxicity; drug resistance to steady repeated therapy and poor drug biodistribution in the targeted area. In brain tumors, these angiogenic inhibitors have limited ability to cross the BBB due to their molecular size, lipophilicity and susceptibility to efflux by P-gp and BCRP. As a result, they have reduced effectiveness in treating brain tumors or brain metastases compared to other malignancies outside the brain. Researchers are continuing to investigate strategies to improve drug delivery to the brain, such as using advanced drug delivery systems, temporary BBB disruption techniques and combination therapies. Despite these difficulties, the development of more efficient angiogenic inhibitors, designed to better cross the BBB, in combination with novel administration methods to evade drug resistance, could encourage significant progress toward treating brain tumors.
Author Contributions
Conceptualization, O.A. and A.D.; software, I.M.B. and L.G.T.; validation, O.A., C.D. and I.S.; formal analysis, O.A., C.D. and I.S.; writing—original draft preparation, O.A., I.M.B. and L.G.T.; writing—review and editing, O.A., C.D., I.S. and A.D.; visualization, L.G.T., C.D. and I.S.; supervision, O.A and A.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grant PN-III-P4-ID-PCE-2020-1649 from the UEFISCDI, Bucharest.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Lei, S.-H. Angiogenic Signaling Pathways and Anti-Angiogenic Therapy for Cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Castellino, R.C.; Durden, D.L. Mechanisms of Disease: The PI3K-Akt-PTEN Signaling Node--an Intercept Point for the Control of Angiogenesis in Brain Tumors. Nat. Clin. Pract. Neurol. 2007, 3, 682–693. [Google Scholar] [CrossRef]
- Zou, X.; Tang, X.Y.; Qu, Z.Y.; Sun, Z.W.; Ji, C.F.; Li, Y.J.; Guo, S.D. Targeting the PDGF/PDGFR Signaling Pathway for Cancer Therapy: A Review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor Signaling Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic Targeting of the Angiopoietin-TIE Pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular Endothelial Growth Factor Induced by Hypoxia May Mediate Hypoxia-Initiated Angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Chaudhry, I.H.; O’Donovan, D.G.; Brenchley, P.E.C.; Reid, H.; Roberts, I.S.D. Vascular Endothelial Growth Factor Expression Correlates with Tumour Grade and Vascularity in Gliomas. Histopathology 2001, 39, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Samoto, K.; Ikezaki, K.; Ono, M.; Shono, T.; Kohno, K.; Kuwano, M.; Fukui, M. Expression of Vascular Endothelial Growth Factor and Its Possible Relation with Neovascularization in Human Brain Tumors. Cancer Res. 1995, 55, 1189–1193. [Google Scholar]
- Auguste, P.; Gürsel, D.B.; Lemière, S.; Reimers, D.; Cuevas, P.; Carceller, F.; Di Santo, J.P.; Bikfalvi, A. Inhibition of Fibroblast Growth Factor/Fibroblast Growth Factor Receptor Activity in Glioma Cells Impedes Tumor Growth by Both Angiogenesis-Dependent and -Independent Mechanisms. Cancer Res. 2001, 61, 1717–1726. [Google Scholar]
- Westermark, B.; Heldin, C.-H.; Nistér, M. Platelet-Derived Growth Factor in Human Glioma. Glia 1995, 15, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H. Mechanisms of Angiogenesis in the Brain. J. Neuropathol. Exp. Neurol. 1999, 58, 313–320. [Google Scholar] [CrossRef]
- Haddad-Tóvolli, R.; Dragano, N.R.V.; Ramalho, A.F.S.; Velloso, L.A. Development and Function of the Blood-Brain Barrier in the Context of Metabolic Control. Front. Neurosci. 2017, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The Blood-Brain Barrier and Blood-Tumour Barrier in Brain Tumours and Metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Hoosemans, L.; Vooijs, M.; Hoeben, A. Opportunities and Challenges of Small Molecule Inhibitors in Glioblastoma Treatment: Lessons Learned from Clinical Trials. Cancers 2024, 16, 3021. [Google Scholar] [CrossRef]
- Zhang, F.; Wen, L.; Wang, K.; Huang, Z.; Jin, X.; Xiong, R.; He, S.; Hu, F. Effect of Axitinib Regulating the Pathological Blood-Brain Barrier Functional Recovery for Glioblastoma Therapeutics. CNS Neurosci. Ther. 2022, 28, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Pagnuzzi-Boncompagni, M.; Picco, V.; Vial, V.; Planas-Bielsa, V.; Vandenberghe, A.; Daubon, T.; Derieppe, M.A.; Montemagno, C.; Durivault, J.; Grépin, R.; et al. Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers 2021, 14, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, W.; Wen, L.; Jin, X.; Meng, T.; Li, S.; Hong, Y.; Xu, Y.; Yuan, H.; Hu, F. The Protective Effects of Axitinib on Blood-Brain Barrier Dysfunction and Ischemia-Reperfusion Injury in Acute Ischemic Stroke. Exp. Neurol. 2024, 379, 114870. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sane, R.; Ohlfest, J.R.; Elmquist, W.F. The Role of the Breast Cancer Resistance Protein (ABCG2) in the Distribution of Sorafenib to the Brain. J. Pharmacol. Exp. Ther. 2011, 336, 223–233. [Google Scholar] [CrossRef]
- Durmus, S.; Xu, N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (MDR1/ABCB1) and Breast Cancer Resistance Protein (BCRP/ABCG2) Restrict Brain Accumulation of the JAK1/2 Inhibitor, CYT387. Pharmacol. Res. 2013, 76, 9–16. [Google Scholar] [CrossRef]
- Oberoi, R.K.; Mittapalli, R.K.; Elmquist, W.F. Pharmacokinetic Assessment of Efflux Transport in Sunitinib Distribution to the Brain. J. Pharmacol. Exp. Ther. 2013, 347, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.M.; Purcaru, S.O.; Alexandru, O.; Dricu, A. New Perspectives in Glioblastoma Antiangiogenic Therapy. Contemp. Oncol. 2016, 20, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination with Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Dasanu, C.A.; Alvarez-Argote, J.; Limonadi, F.M.; Codreanu, I. Bevacizumab in Refractory Higher-Grade and Atypical Meningioma: The Current State of Affairs. Expert Opin. Biol. Ther. 2019, 19, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, X.; Wang, H.; Chen, G.; Zhou, Y. Anti-VEGFR2 Monoclonal Antibody(MSB0254) Inhibits Angiogenesis and Tumor Growth by Blocking the Signaling Pathway Mediated by VEGFR2 in Glioblastoma. Biochem. Biophys. Res. Commun. 2022, 604, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Cher, L.; Nowak, A.; Iatropoulos, G.; Lee, W.S.; Lee, S.Y.; Shim, S.R.; Yoo, J.S. ACTR-75. A multicenter, 3-ARM, open-label, phase IIA clinical trial to evaluate safety and efficacy of tanibirumab (VEGFR2 MAB), in patients with recurrent GBM assessed with K-trans and initial area under the gadolinium concentration-time curve (IAUGC). Neuro Oncol. 2017, 19, vi17. [Google Scholar] [CrossRef]
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Tache, D.E.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the VEGF and PDGF Signaling Pathway in Glioblastoma Treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837. [Google Scholar] [PubMed]
- Bergers, G.; Hanahan, D. Modes of Resistance to Anti-Angiogenic Therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef]
- Serban, F.; Daianu, O.; Tataranu, L.G.; Artene, S.A.; Emami, G.; Georgescu, A.M.; Alexandru, O.; Purcaru, S.O.; Tache, D.E.; Danciulescu, M.M.; et al. Silencing of Epidermal Growth Factor, Latrophilin and Seven Transmembrane Domain-Containing Protein 1 (ELTD1) via SiRNA-Induced Cell Death in Glioblastoma. J. Immunoass. Immunochem. 2017, 38, 21–33. [Google Scholar] [CrossRef]
- Sevastre, A.S.; Buzatu, I.M.; Baloi, C.; Oprita, A.; Dragoi, A.; Tataranu, L.G.; Alexandru, O.; Tudorache, S.; Dricu, A. ELTD1—An Emerging Silent Actor in Cancer Drama Play. Int. J. Mol. Sci. 2021, 22, 5151. [Google Scholar] [CrossRef]
- Tataranu, L.G.; Turliuc, S.; Rizea, R.E.; Dricu, A.; Alexandru, O.; Staicu, G.A.; Kamel, A. A Synopsis of Biomarkers in Glioblastoma: Past and Present. Curr. Issues Mol. Biol. 2024, 46, 6903–6939. [Google Scholar] [CrossRef]
- Serban, F.; Artene, S.A.; Georgescu, A.M.; Purcaru, S.O.; Tache, D.E.; Alexandru, O.; Dricu, A. Epidermal Growth Factor, Latrophilin, and Seven Transmembrane Domain-Containing Protein 1 Marker, a Novel Angiogenesis Marker. Onco Targets Ther. 2015, 8, 3767. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Kessler, S.M.; Kumar, V.; Zwergel, C. Editorial: Multi-Target Directed Ligands for the Treatment of Cancer. Front. Oncol. 2022, 12, 980141. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- US Food and Drug Administration FDA Commissioner Announces Avastin Decision|Fierce Pharma. Available online: https://www.fiercepharma.com/pharma/fda-commissioner-announces-avastin-decision-0 (accessed on 6 January 2025).
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic Therapy in Oncology: Current Status and Future Directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-Angiogenic Agents for the Treatment of Solid Tumors: Potential Pathways, Therapy and Current Strategies—A Review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Lisi, L.; Bonanno, E.; Scimeca, M.; Lagana, A.; Barca, S.; Caputo, M.; et al. The Anti-Vascular Endothelial Growth Factor Receptor-1 Monoclonal Antibody D16F7 Inhibits Invasiveness of Human Glioblastoma and Glioblastoma Stem Cells. J. Exp. Clin. Cancer Res. 2017, 36, 106. [Google Scholar] [CrossRef]
- Jones, K.A.; Reeves, R.; Gupta, A.; Piao, Y.; Li, S.; Lin, S.-Y.; Hu, Y. Selective Coexpression of VEGF Receptor 2 in EGFRvIII-Positive Glioblastoma Cells Prevents Cellular Senescence and Contributes to Their Aggressive Nature. Neuro Oncol. 2016, 18, 667–678. [Google Scholar] [CrossRef]
- Grau, S.J.; Trillsch, F.; Tonn, J.C.; Goldbrunner, R.H.; Noessner, E. Expression of VEGFR3 in Glioma Endothelium Correlates with Tumor Grade. J. Neurooncol. 2007, 82, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Sasagawa, Y.; Tachibana, O.; Iizuka, H.; Kurose, N.; Shioya, A.; Guo, X.; Yamada, S.; Nojima, T. The Clinicopathological Analysis of Receptor Tyrosine Kinases in Meningiomas: The Expression of VEGFR-2 in Meningioma Was Associated with a Higher WHO Grade and Shorter Progression-Free Survival. Brain Tumor Pathol. 2019, 36, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO Guidelines on the Diagnosis and Treatment of Diffuse Gliomas of Adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF Receptors in Glioma. Upsala J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef]
- Guérit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.-B. PDGF Receptor Mutations in Human Diseases. Cell. Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Yin, S.; Yang, S.; Jiang, X.; Wang, J.; Zhang, M.; Zhang, L. Roles of Platelets in Tumor Invasion and Metastasis: A Review. Heliyon 2022, 8, e12072. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Puputti, M.; Sihto, H.; Tynninen, O.; Nupponen, N.N. Amplification of Genes Encoding KIT, PDGFRalpha and VEGFR2 Receptor Tyrosine Kinases Is Frequent in Glioblastoma Multiforme. J. Pathol. 2005, 207, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef]
- Puputti, M.; Tynninen, O.; Sihto, H.; Blom, T.; Mäenpää, H.; Isola, J.; Paetau, A.; Joensuu, H.; Nupponen, N.N. Amplification of KIT, PDGFRA, VEGFR2, and EGFR in Gliomas. Mol. Cancer Res. 2006, 4, 927–934. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, G. Genetic Alterations and Aberrant Expression of Genes Related to the Phosphatidyl-Inositol-3’-Kinase/Protein Kinase B (Akt) Signal Transduction Pathway in Glioblastomas. Brain Pathol. 2003, 13, 507–518. [Google Scholar] [CrossRef]
- Peng, G.; Wang, Y.; Ge, P.; Bailey, C.; Zhang, P.; Zhang, D.; Meng, Z.; Qi, C.; Chen, Q.; Chen, J.; et al. The HIF1α-PDGFD-PDGFRα Axis Controls Glioblastoma Growth at Normoxia/Mild-Hypoxia and Confers Sensitivity to Targeted Therapy by Echinomycin. J. Exp. Clin. Cancer Res. 2021, 40, 278. [Google Scholar] [CrossRef]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant Glioma: Genetics and Biology of a Grave Matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Carapancea, M.; Cosaceanu, D.; Budiu, R.; Kwiecinska, A.; Tataranu, L.; Ciubotaru, V.; Alexandru, O.; Banita, M.; Pisoschi, C.; Bäcklund, M.L.; et al. Dual Targeting of IGF-1R and PDGFR Inhibits Proliferation in High-Grade Gliomas Cells and Induces Radiosensitivity in JNK-1 Expressing Cells. J. Neurooncol. 2007, 85, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Carapancea, M.; Alexandru, O.; Fetea, A.S.; Dragutescu, L.; Castro, J.; Georgescu, A.; Popa-Wagner, A.; Bäcklund, M.L.; Lewensohn, R.; Dricu, A. Growth Factor Receptors Signaling in Glioblastoma Cells: Therapeutic Implications. J. Neurooncol. 2009, 92, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.-H.; Westermark, B.; Nistér, M. Platelet-Derived Growth Factor and Its Receptors in Human Glioma Tissue: Expression of Messenger RNA and Protein Suggests the Presence of Autocrine and Paracrine Loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Black, P.M.; Carroll, R.; Glowacka, D.; Riley, K.; Dashner, K. Platelet-Derived Growth Factor Expression and Stimulation in Human Meningiomas. J. Neurosurg. 1994, 81, 388–393. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast Growth Factor Receptors as Treatment Targets in Clinical Oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; Lucks, P.; Borghouts, C. The Function of Stat3 in Tumor Cells and Their Microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef]
- Toyoda, K.; Oishi, H.; Shimizu, K.; Miyamoto, S.; Kuroda, S.; Hida, K. Initial Contact of Glioblastoma Cells with Existing Normal Brain Endothelial Cells Strengthens the Barrier Function via Fibroblast Growth Factor 2 Secretion: A New in Vitro Blood–Brain Barrier Model. Cell. Mol. Neurobiol. 2013, 33, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Stan, A.C.; Nemati, M.N.; Pietsch, T.; Hüll, M.; Graf, R.; Seifert, V. In Vivo Inhibition of Angiogenesis and Growth of the Human U-87 Malignant Glial Tumor by Treatment with an Antibody against Basic Fibroblast Growth Factor. J. Neurosurg. 1995, 82, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Lamszus, K.; Lengler, U.; Schmidt, N.O.; Stavrou, D.; Ergün, S.; Westphal, M. Vascular Endothelial Growth Factor, Hepatocyte Growth Factor/Scatter Factor, Basic Fibroblast Growth Factor, and Placenta Growth Factor in Human Meningiomas and Their Relation to Angiogenesis and Malignancy. Neurosurgery 2000, 46, 938–948. [Google Scholar] [CrossRef]
- Dumont, D.J.; Yamaguchi, T.P.; Conlon, R.A.; Rossant, J.; Breitman, M.L. Tek, a Novel Tyrosine Kinase Gene Located on Mouse Chromosome 4, Is Expressed in Endothelial Cells and Their Presumptive Precursors. Oncogene 1992, 7, 1471–1480. [Google Scholar]
- Fukuhara, S.; Sako, K.; Minami, T.; Noda, K.; Kim, H.-R.; Kodama, T.; Shibuya, M.; Takakura, N.; Koh, G.Y.; Mochizuki, N. Differential Function of Tie2 at Cell–Cell Contacts and Cell–Substratum Contacts Regulated by Angiopoietin-1. Nat. Cell Biol. 2008, 10, 513–526. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Pulkki, K.; Bono, P.; Alitalo, K. Angiopoietins Assemble Distinct Tie2 Signalling Complexes in Endothelial Cell–Cell and Cell–Matrix Contacts. Nat. Cell Biol. 2008, 10, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Oike, Y.; Akao, M.; Kubota, Y.; Suda, T. Angiopoietin-like Proteins: Potential New Targets for Metabolic Syndrome Therapy. Trends. Mol. Med. 2005, 11, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Liu, L.; Wang, Z.; Cai, H.; Hu, W.; Jia, W.; Yuan, Z.; Yuan, J.; Zhong, T. Viral G Protein-Coupled Receptor up-Regulates Angiopoietin-like 4 Promoting Angiogenesis and Vascular Permeability in Kaposi’s Sarcoma. Proc. Natl. Acad. Sci. USA 2010, 107, 14363–14368. [Google Scholar] [CrossRef] [PubMed]
- Brunckhorst, M.K.; Wang, H.; Lu, R.; Yu, Q. Angiopoietin-4 Promotes Glioblastoma Progression by Enhancing Tumor Cell Viability and Angiogenesis. Cancer Res. 2010, 70, 7283–7293. [Google Scholar] [CrossRef] [PubMed]
- Audero, E.; Cascone, I.; Zanon, I.; Previtali, S.C.; Piva, R.; Schiffer, D.; Bussolino, F. Expression of Angiopoietin-1 in Human Glioblastomas Regulates Tumor-Induced Angiogenesis: In Vivo and in Vitro Studies. Arter. Thromb. Vasc. Biol. 2001, 21, 536–541. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Ilhan, A.; Gartner, W.; Neziri, D.; Czech, T.; Base, W.; Hörl, W.H.; Wagner, L. Angiogenic Factors in Plasma of Brain Tumour Patients. Anticancer Res. 2009, 29, 731–736. [Google Scholar]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative Effectiveness of Axitinib versus Sorafenib in Advanced Renal Cell Carcinoma (AXIS): A Randomised Phase 3 Trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Tiako, M.J.; Lebrun-Vignes, B.; Martin, M.; Zohar, S.; Salem, J.-E. A Profile of Avelumab plus Axitinib in the Treatment of Renal Cell Carcinoma. Ther. Clin. Risk Manag. 2022, 18, 683–698. [Google Scholar] [CrossRef]
- Kania, R.S. Structure-Based Design and Characterization of Axitinib. In Kinase Inhibitor Drugs; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 167–201. [Google Scholar]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A.; ESMO Guidelines Committee. Renal Cell Carcinoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef]
- Chen, Y.; Tortorici, M.A.; Garrett, M.; Hee, B.; Fujiwara, Y.; Mulay, M. Clinical Pharmacology of Axitinib. Clin. Pharmacokinet. 2013, 52, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Inc. INLYTA® (Axitinib) Tablets for Oral. Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202324lbl.pdf (accessed on 2 December 2024).
- Cohen, E.E.W.; Rosen, L.S.; Vokes, E.E.; Kies, M.S.; Forastiere, A.A.; Worden, F.P.; Kane, M.A.; Sherman, S.I.; Kim, S.-J.; Bycott, P.W.; et al. Axitinib Is an Active Treatment for All Histologic Subtypes of Advanced Thyroid Cancer: Results from a Phase II Study. J. Clin. Oncol. 2008, 26, 4708–4713. [Google Scholar] [CrossRef]
- Paik, E.S.; Kim, T.-H.; Cho, Y.J.; Ryu, J.; Choi, J.-J.; Lee, Y.-Y.; Kim, T.-J.; Choi, C.-H.; Kim, W.Y.; Sa, J.K.; et al. Preclinical Assessment of the VEGFR Inhibitor Axitinib as a Therapeutic Agent for Epithelial Ovarian Cancer. Sci. Rep. 2020, 10, 4904. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Ma, B.B.Y.; Loong, H.H.F.; Mo, F.; Li, L.; King, A.D.; Wang, K.; Ahuja, A.T.; Chan, C.M.L.; Hui, C.W.C.; et al. Efficacy, Safety, and Pharmacokinetics of Axitinib in Nasopharyngeal Carcinoma: A Preclinical and Phase II Correlative Study. Clin. Cancer Res. 2018, 24, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-Z.; Lin, C.-H.; Wang, Y.-J.; Chao, Y.; Chiou, J.-Y.; Yen, C.-J.; Lee, K.-D.; Wang, C.-K.; Chang, J.W.C.; Huang, C.-T. A Multicenter Phase II Study of Second-Line Axitinib for Patients with Advanced Hepatocellular Carcinoma Failing First-Line Sorafenib Monotherapy. Oncologist 2020, 25, e1280–e1285. [Google Scholar] [CrossRef]
- Schiller, J.H.; Larson, T.; Ou, S.-H.I.; Golshan, F.; Mandrekar, S.J.; Carbone, D.P.; Li, S.; Bair, A.H.; Knost, J.A. Efficacy and Safety of Axitinib in Patients with Advanced Non–Small-Cell Lung Cancer: Results from a Phase II Study. J. Clin. Oncol. 2009, 27, 3836–3841. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Lutzky, J.; McDermott, D.F.; Brown, C.K.; Meric, J.B.; Rosbrook, B.; Wang, J.; Kiedrowski, T.; Gulyas, S.; Sandor, V. Multicenter, Phase II Study of Axitinib, a Selective Second-Generation Inhibitor of Vascular Endothelial Growth Factor Receptors 1, 2, and 3, in Patients with Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 7462–7469. [Google Scholar] [CrossRef]
- Wilmes, L.J.; Pallavicini, M.G.; Fleming, L.M.; Gibbs, J.; Wang, D.; Li, K.-L.; Partridge, S.C.; Henry, R.G.; Shalinsky, D.R.; Hu-Lowe, D.; et al. AG-013736, a Novel Inhibitor of VEGF Receptor Tyrosine Kinases, Inhibits Breast Cancer Growth and Decreases Vascular Permeability as Detected by Dynamic Contrast-Enhanced Magnetic Resonance Imaging. Magn. Reson. Imaging 2007, 25, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Grávalos, C.; Carrato, A.; Tobeña, M.; Rodriguez-Garrote, M.; Soler, G.; Vieitez, J.M.; Robles, L.; Valladares-Ayerbes, M.; Polo, E.; Limón, M.L.; et al. A Randomized Phase II Study of Axitinib as Maintenance Therapy After First-Line Treatment for Metastatic Colorectal Cancer. Clin. Color. Cancer 2018, 17, e323–e329. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saha, D.; Martuza, R.L.; Rabkin, S.D.; Wakimoto, H. Single Agent Efficacy of the VEGFR Kinase Inhibitor Axitinib in Preclinical Models of Glioblastoma. J. Neurooncol. 2015, 121, 91–100. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Cardinali, C.; Santoni, M.; Bernardini, G.; Santoni, A.; Santoni, G. Axitinib Induces Senescence-Associated Cell Death and Necrosis in Glioma Cell Lines. Oncotarget 2017, 8, 2632–2643. [Google Scholar] [CrossRef]
- Krcek, R.; Matschke, V.; Theis, V.; Adamietz, I.A.; Bühler, H.; Theiss, C. Vascular Endothelial Growth Factor, Irradiation, and Axitinib Have Diverse Effects on Motility and Proliferation of Glioblastoma Multiforme Cells. Front. Oncol. 2017, 7, 182. [Google Scholar] [CrossRef]
- Opriţa, A.; Dobrescu, M.A.; Manea, E.V.; Popescu, Ş.O.; Sevastre, A.S.; Pîrvu, A.S.; Buzatu, I.M.; Tache, D.E. In Vitro Evaluation of Axitinib and Sorafenib Treatment in Glioblastoma Cell Viability and Morphology. Rom. J. Morphol. Embryol. 2023, 64, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef]
- Schwinn, S.; Mokhtari, Z.; Thusek, S.; Schneider, T.; Sirén, A.-L.; Tiemeyer, N.; Caruana, I.; Miele, E.; Schlegel, P.G.; Beilhack, A.; et al. Cytotoxic Effects and Tolerability of Gemcitabine and Axitinib in a Xenograft Model for C-Myc Amplified Medulloblastoma. Sci. Rep. 2021, 11, 14062. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Freitag, D.; Grube, S.; Dunisch, P.; Kalff, R.; Ewald, C. Multi Tyrosin Kinase Inihibition by Axitinib in the Treatment of Human Meningiomas. In Proceedings of the Deutsche Gesellschaft für Neurochirurgie; German Medical Science GMS Publishing House, Düsseldorf, Germany, 26–29 May 2013. [Google Scholar]
- Bayer NEXAVAR (Sorafenib) Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021923s020lbl.pdf (accessed on 2 December 2024).
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.-M.; Zhang, X.; Vincent, P.; McHugh, M. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the Raf/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Hendlisz, A.; Gil, T.; Bartholomeus, S.; Mano, M.; de Valeriola, D.; Strumberg, D.; Brendel, E.; Haase, C.G.; Schwartz, B.; et al. Phase I Safety and Pharmacokinetics of BAY 43-9006 Administered for 21 Days on/7 Days off in Patients with Advanced, Refractory Solid Tumours. Br. J. Cancer 2005, 92, 1855–1861. [Google Scholar] [CrossRef]
- Yu, C.; Friday, B.B.; Lai, J.-P.; Yang, L.; Sarkaria, J.; Kay, N.E.; Carter, C.A.; Roberts, L.R.; Kaufmann, S.H.; Adjei, A.A. Cytotoxic Synergy between the Multikinase Inhibitor Sorafenib and the Proteasome Inhibitor Bortezomib in Vitro: Induction of Apoptosis through Akt and c-Jun NH2-Terminal Kinase Pathways. Mol. Cancer Ther. 2006, 5, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Raskett, C.M.; Gilbert, C.A.; Ross, A.H.; Altieri, D.C. Sorafenib Exerts Anti-Glioma Activity in Vitro and in Vivo. Neurosci. Lett. 2010, 478, 165–170. [Google Scholar] [CrossRef]
- Yang, F.; Brown, C.; Buettner, R.; Hedvat, M.; Starr, R.; Scuto, A.; Schroeder, A.; Jensen, M.; Jove, R. Sorafenib Induces Growth Arrest and Apoptosis of Human Glioblastoma Cells through the Dephosphorylation of Signal Transducers and Activators of Transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib Selectively Depletes Human Glioblastoma Tumor-Initiating Cells from Primary Cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef]
- Riedel, M.; Struve, N.; Müller-Goebel, J.; Köcher, S.; Petersen, C.; Dikomey, E.; Rothkamm, K.; Kriegs, M. Sorafenib Inhibits Cell Growth but Fails to Enhance Radio- and Chemosensitivity of Glioblastoma Cell Lines. Oncotarget 2016, 7, 61988–61995. [Google Scholar] [CrossRef]
- Wilisch-Neumann, A.; Pachow, D.; Kirches, E.; Mawrin, C. MS-29: Sorafenib and regorafenib inhibit growth and migration of meningioma cells. Neuro Oncol. 2014, 16, v132. [Google Scholar] [CrossRef][Green Version]
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor Receptor (VEGF-R) 2 and VEGF-R3 Kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [Google Scholar] [CrossRef]
- Glen, H.; Mason, S.; Patel, H.; Nathubhai, A.; Muralikrishnan, S.; Pillai, M. E7080, a Multi-Targeted Tyrosine Kinase Inhibitor Suppresses Tumor Cell Migration and Invasion. BMC Cancer 2011, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, R.; Aluri, J.; Fan, J.; Martinez, G.; Pentikis, H.; Ren, M. Influence of Hepatic Impairment on Lenvatinib Pharmacokinetics Following Single-Dose Oral Administration. J. Clin. Pharmacol. 2015, 55, 317–327. [Google Scholar] [CrossRef]
- Dubbelman, A.-C.; Rosing, H.; Nijenhuis, C.; Huitema, A.D.R.; Mergui-Roelvink, M.; Gupta, A.; Verbel, D.; Thompson, G.; Shumaker, R.; Schellens, J.H.M.; et al. Pharmacokinetics and Excretion of (14)C-Lenvatinib in Patients with Advanced Solid Tumors or Lymphomas. Investig. New Drugs 2015, 33, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Yamamoto, Y.; Funahashi, Y.; Tsuruoka, A.; Watanabe, T.; Wakabayashi, T.; Uenaka, T.; Asada, M. E7080, a Novel Inhibitor That Targets Multiple Kinases, Has Potent Antitumor Activities against Stem Cell Factor Producing Human Small Cell Lung Cancer H146, Based on Angiogenesis Inhibition. Int. J. Cancer 2008, 122, 664–671. [Google Scholar] [CrossRef]
- Ikuta, K.; Yano, S.; Trung, V.T.; Hanibuchi, M.; Goto, H.; Li, Q.; Wang, W.; Yamada, T.; Ogino, H.; Kakiuchi, S.; et al. E7080, a Multi-Tyrosine Kinase Inhibitor, Suppresses the Progression of Malignant Pleural Mesothelioma with Different Proangiogenic Cytokine Production Profiles. Clin. Cancer Res. 2009, 15, 7229–7237. [Google Scholar] [CrossRef]
- Li, J.; Zou, C.-L.; Zhang, Z.-M.; Lv, L.-J.; Qiao, H.-B.; Chen, X.-J. A Multi-targeted Tyrosine Kinase Inhibitor Lenvatinib for the Treatment of Mice with Advanced Glioblastoma. Mol. Med. Rep. 2017, 16, 7105–7111. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Pazopanib Clinical Pharmacology and Biopharmaceutics Review. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_ClinPharmR.pdf (accessed on 2 December 2024).
- Novartis VOTRIENT® (Pazopanib) Tablets, for Oral Use. Available online: https://www.novartis.com/us-en/sites/novartis_us/files/votrient.pdf (accessed on 2 December 2024).
- Imbs, D.-C.; Paludetto, M.-N.; Négrier, S.; Powell, H.; Lafont, T.; White-Koning, M.; Chatelut, E.; Thomas, F. Determination of Unbound Fraction of Pazopanib in Vitro and in Cancer Patients Reveals Albumin as the Main Binding Site. Investig. New Drugs 2016, 34, 41–48. [Google Scholar] [CrossRef]
- European Medicines Agency Votrient® (Pazopanib): Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/votrient-epar-product-information_en.pdf (accessed on 29 April 2012).
- Deng, Y.; Sychterz, C.; Suttle, A.B.; Dar, M.M.; Bershas, D.; Negash, K.; Qian, Y.; Chen, E.P.; Gorycki, P.D.; Ho, M.Y.K. Bioavailability, Metabolism and Disposition of Oral Pazopanib in Patients with Advanced Cancer. Xenobiotica 2013, 43, 443–453. [Google Scholar] [CrossRef]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.-C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-Pharmacodynamic Correlation from Mouse to Human with Pazopanib, a Multikinase Angiogenesis Inhibitor with Potent Antitumor and Antiangiogenic Activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef]
- Pfizer Inc. SUTENT- Sunitinib Malate Capsule. Available online: https://labeling.pfizer.com/ShowLabeling.aspx?id=607 (accessed on 6 January 2025).
- Kassem, M.G.; Motiur Rahman, A.F.M.; Korashy, H.M. Sunitinib Malate. Profiles Drug Subst. Excip. Relat. Methodol. 2012, 37, 363–388. [Google Scholar] [CrossRef] [PubMed]
- De Boüard, S.; Herlin, P.; Christensen, J.G.; Lemoisson, E.; Gauduchon, P.; Raymond, E.; Guillamo, J.S. Antiangiogenic and Anti-Invasive Effects of Sunitinib on Experimental Human Glioblastoma. Neuro Oncol. 2007, 9, 412–423. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Lei, L.; Kennedy, B.C.; Sisti, J.; Ebiana, V.; Crisman, C.; Christensen, J.G.; Gil, O.; Rosenfeld, S.S.; Canoll, P.; et al. The Addition of Sunitinib to Radiation Delays Tumor Growth in a Murine Model of Glioblastoma. Neurol. Res. 2012, 34, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The Brain-Penetrating CXCR4 Antagonist, PRX177561, Increases the Antitumor Effects of Bevacizumab and Sunitinib in Preclinical Models of Human Glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Dabkeviciute, G.; Maccioni, E.; Petrikaite, V. Effect of Sunitinib Derivatives on Glioblastoma Single-Cell Migration and 3D Cell Cultures. Am. J. Cancer Res. 2023, 13, 1377–1386. [Google Scholar] [PubMed]
- Ho, K.H.; Lee, Y.T.; Chen, P.H.; Shih, C.M.; Cheng, C.H.; Chen, K.C. Guanabenz Sensitizes Glioblastoma Cells to Sunitinib by Inhibiting GADD34-Mediated Autophagic Signaling. Neurotherapeutics 2021, 18, 1371–1392. [Google Scholar] [CrossRef] [PubMed]
- Andrae, N.; Kirches, E.; Hartig, R.; Haase, D.; Keilhoff, G.; Kalinski, T.; Mawrin, C. Sunitinib Targets PDGF-Receptor and Flt3 and Reduces Survival and Migration of Human Meningioma Cells. Eur. J. Cancer 2012, 48, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.J.; Rhodes, S.D.; Jiang, L.; Li, X.; Yuan, J.; Yang, X.; Zhang, S.; Vakili, S.T.; Territo, P.; Hutchins, G.; et al. Preclinical Evidence for the Use of Sunitinib Malate in the Treatment of Plexiform Neurofibromas. Pediatr. Blood Cancer 2016, 63, 206–213. [Google Scholar] [CrossRef]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Holland, J.; Ramies, D.; Mamelok, R.; Benrimoh, N.; Ciric, S.; Marbury, T.; Preston, R.A.; Heuman, D.M.; Gavis, E.; et al. Effect of Renal and Hepatic Impairment on the Pharmacokinetics of Cabozantinib. J. Clin. Pharmacol. 2016, 56, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Bentzien, F.; Zuzow, M.; Heald, N.; Gibson, A.; Shi, Y.; Goon, L.; Yu, P.; Engst, S.; Zhang, W.; Huang, D.; et al. In Vitro and in Vivo Activity of Cabozantinib (XL184), an Inhibitor of RET, MET, and VEGFR2, in a Model of Medullary Thyroid Cancer. Thyroid 2013, 23, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Swanson, K.D.; Csizmadia, E.; Solanki, A.; Landon-Brace, N.; Gehring, M.P.; Helenius, K.; Olson, B.M.; Pyzer, A.R.; Wang, L.C.; et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017, 7, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Fioramonti, M.; Fausti, V.; Pantano, F.; Iuliani, M.; Ribelli, G.; Lotti, F.; Pignochino, Y.; Grignani, G.; Santini, D.; Tonini, G.; et al. Cabozantinib Affects Osteosarcoma Growth Through A Direct Effect On Tumor Cells and Modifications In Bone Microenvironment. Sci. Rep. 2018, 8, 4177. [Google Scholar] [CrossRef]
- Fuse, M.A.; Plati, S.K.; Burns, S.S.; Dinh, C.T.; Bracho, O.; Yan, D.; Mittal, R.; Shen, R.; Soulakova, J.N.; Copik, A.J.; et al. Combination Therapy with C-Met and Src Inhibitors Induces Caspase-Dependent Apoptosis of Merlin-Deficient Schwann Cells and Suppresses Growth of Schwannoma Cells. Mol. Cancer Ther. 2017, 16, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Gebreyohannes, Y.K.; Schöffski, P.; Van Looy, T.; Wellens, J.; Vreys, L.; Cornillie, J.; Vanleeuw, U.; Aftab, D.T.; Debiec-Rychter, M.; Sciot, R.; et al. Cabozantinib Is Active against Human Gastrointestinal Stromal Tumor Xenografts Carrying Different KIT Mutations. Mol. Cancer Ther. 2016, 15, 2845–2852. [Google Scholar] [CrossRef]
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schönsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The Novel C-Met Inhibitor Cabozantinib Overcomes Gemcitabine Resistance and Stem Cell Signaling in Pancreatic Cancer. Cell Death Dis. 2013, 4, e627. [Google Scholar] [CrossRef]
- Navis, A.C.; Bourgonje, A.; Wesseling, P.; Wright, A.; Hendriks, W.; Verrijp, K.; van der Laak, J.A.W.M.; Heerschap, A.; Leenders, W.P.J. Effects of Dual Targeting of Tumor Cells and Stroma in Human Glioblastoma Xenografts with a Tyrosine Kinase Inhibitor against C-MET and VEGFR2. PLoS ONE 2013, 8, e58262. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Tonse, R.; Appel, H.; Odia, Y.; Kotecha, R.R.; Rabinowits, G.; Mehta, M.P. Regression of Intracranial Meningiomas Following Treatment with Cabozantinib. Curr. Oncol. 2021, 28, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Apoverlag MeinAPOVERLAG—Austria-Codex Online. Available online: https://mein.apoverlag.at/austriacodex (accessed on 6 January 2025).
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Gabasa, M.; Ikemori, R.; Hilberg, F.; Reguart, N.; Alcaraz, J. Nintedanib Selectively Inhibits the Activation and Tumour-Promoting Effects of Fibroblasts from Lung Adenocarcinoma Patients. Br. J. Cancer 2017, 117, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Sagara, A.; Fukuchi, Y.; Muto, A. Single-agent Nintedanib Suppresses Metastatic Osteosarcoma Growth by Inhibiting Tumor Vascular Formation. Oncol. Lett. 2024, 27, 123. [Google Scholar] [CrossRef]
- Da Silva, R.F.; Nogueira-Pangrazi, E.; Kido, L.A.; Montico, F.; Arana, S.; Kumar, D.; Raina, K.; Agarwal, R.; Cagnon, V.H.A. Nintedanib Antiangiogenic Inhibitor Effectiveness in Delaying Adenocarcinoma Progression in Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP). J. Biomed. Sci. 2017, 24, 31. [Google Scholar] [CrossRef]
- Török, S.; Cserepes, T.M.; Rényi-Vámos, F.; Döme, B. Nintedanib (BIBF 1120) in the Treatment of Solid Cancers: An Overview of Biological and Clinical Aspects. Magy Onkol. 2012, 56, 199–208. [Google Scholar] [PubMed]
- Dang, Y.; Zhao, Z.; Wang, B.; Du, A.; Li, S.; Yuan, G.; Pan, Y. Polymeric Polylactic Acid-Glycolic Acid-Based Nanoparticles Deliver Nintedanib Across the Blood-Brain Barrier to Inhibit Glioblastoma Growth. Int. J. Mol. Sci. 2025, 26, 443. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration, Stivarga® (Regorafenib) [Prescribing Information. 2015]. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/203085s004lbl.pdf (accessed on 27 April 2017).
- Parsad, S.; Ratain, M.J. Food Effect Studies for Oncology Drug Products. Clin. Pharmacol. Ther. 2017, 101, 606–612. [Google Scholar] [CrossRef]
- Keunecke, A.; Hoefman, S.; Drenth, H.J.; Zisowsky, J.; Cleton, A.; Ploeger, B.A. Population Pharmacokinetics of Regorafenib in Solid Tumours: Exposure in Clinical Practice Considering Enterohepatic Circulation and Food Intake. Br. J. Clin. Pharmacol. 2020, 86, 2362–2376. [Google Scholar] [CrossRef]
- Subramonian, D.; Phanhthilath, N.; Rinehardt, H.; Flynn, S.; Huo, Y.; Zhang, J.; Messer, K.; Mo, Q.; Huang, S.; Lesperance, J.; et al. Regorafenib Is Effective against Neuroblastoma in Vitro and in Vivo and Inhibits the RAS/MAPK, PI3K/Akt/MTOR and Fos/Jun Pathways. Br. J. Cancer 2020, 123, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wu, L.W.; Zhang, Z.Y.; Chen, M.L.; Li, Y.L.; Zhang, C. The Anti-Tumor Effect of Regorafenib in Lung Squamous Cell Carcinoma in Vitro. Biochem. Biophys. Res. Commun. 2018, 503, 1123–1129. [Google Scholar] [CrossRef]
- Munoz Garcia, J.; Vargas-Franco, J.W.; Bompas, E.; Cochonneau, D.; Ollivier, E.; Kerzerho, J.; Brahmi, M.; Blay, J.-Y.; Heymann, M.-F.; Lezot, F.; et al. Effect of Dual Properties of Regorafenib in Osteosarcoma on Tumor Progression and Bone Parameters in Mouse Preclinical Models. J. Clin. Oncol. 2023, 41, e23501. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A New Oral Multikinase Inhibitor of Angiogenic, Stromal and Oncogenic Receptor Tyrosine Kinases with Potent Preclinical Antitumor Activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Deshors, P.; Arnauduc, F.; Boëlle, B.; Cohen-Jonathan Moyal, E.; Courtade-Saïdi, M.; Evrard, S.M. Impact of Regorafenib on Endothelial Transdifferentiation of Glioblastoma Stem-like Cells. Cancers 2022, 14, 1551. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Buccarelli, M.; Formato, A.; Orecchini, E.; Salbini, M.; Ricci, V.; Orsini, T.; Putti, S.; Chiesa, S.; Ricci-Vitiani, L.; et al. Characterization of Glioblastoma Cells Response to Regorafenib. Cancers 2022, 14, 6193. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, L.; Chen, H.; Lei, Y.; Zhang, T.; Wang, Y.; Jin, P.; Lan, J.; Zhou, L.; Huang, Z.; et al. Regorafenib Induces Lethal Autophagy Arrest by Stabilizing PSAT1 in Glioblastoma. Autophagy 2020, 16, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Chiang, I.T.; Liu, Y.C.; Liu, H.S.; Ali, A.A.A.; Chou, S.Y.; Hsu, T.I.; Hsu, F.T. Regorafenib Reverses Temozolomide-Induced CXCL12/CXCR4 Signaling and Triggers Apoptosis Mechanism in Glioblastoma. Neurotherapeutics 2022, 19, 616–634. [Google Scholar] [CrossRef] [PubMed]
- Tuchen, M.; Wilisch-Neumann, A.; Daniel, E.A.; Baldauf, L.; Pachow, D.; Scholz, J.; Angenstein, F.; Stork, O.; Kirches, E.; Mawrin, C. Receptor Tyrosine Kinase Inhibition by Regorafenib/Sorafenib Inhibits Growth and Invasion of Meningioma Cells. Eur. J. Cancer 2017, 73, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. Aflibercept (VEGF-TRAP): The next Anti-VEGF Drug. Inflamm. Allergy Drug Targets 2011, 10, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. Extended Duration Vascular Endothelial Growth Factor Inhibition in the Eye: Failures, Successes, and Future Possibilities. Pharmaceutics 2018, 10, 21. [Google Scholar] [CrossRef]
- Kim, D.Y.; Choi, J.A.; Koh, J.Y.; Yoon, Y.H. Efficacy and Safety of Aflibercept in in Vitro and in Vivo Models of Retinoblastoma. J. Exp. Clin. Cancer Res. 2016, 35, 171. [Google Scholar] [CrossRef]
- Chiron, M.; Bagley, R.G.; Pollard, J.; Mankoo, P.K.; Henry, C.; Vincent, L.; Geslin, C.; Baltes, N.; Bergstrom, D.A. Differential Antitumor Activity of Aflibercept and Bevacizumab in Patient-Derived Xenograft Models of Colorectal Cancer. Mol. Cancer Ther. 2014, 13, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Wachsberger, P.R.; Burd, R.; Cardi, C.; Thakur, M.; Daskalakis, C.; Holash, J.; Yancopoulos, G.D.; Dicker, A.P. VEGF Trap in Combination with Radiotherapy Improves Tumor Control in U87 Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1526–1537. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomez-Manzano, C.; Holash, J.; Fueyo, J.; Xu, J.; Conrad, C.A.; Aldape, K.D.; De Groot, J.F.; Ekele, B.N.; Yung, W.K.A. VEGF Trap Induces Antiglioma Effect at Different Stages of Disease. Neuro Oncol. 2008, 10, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Eli Lilly and Company Prescribing Information for CYRAMZA (Ramucirumab) Injection, for Intravenous UseInitial U.S. Approval: 2014. Available online: https://uspl.lilly.com/cyramza/cyramza.html#pi (accessed on 6 January 2025).
- Spratlin, J.L.; Cohen, R.B.; Eadens, M.; Gore, L.; Camidge, D.R.; Diab, S.; Leong, S.; O’Bryant, C.; Chow, L.Q.M.; Serkova, N.J.; et al. Phase I Pharmacologic and Biologic Study of Ramucirumab (IMC-1121B), a Fully Human Immunoglobulin G1 Monoclonal Antibody Targeting the Vascular Endothelial Growth Factor Receptor-2. J. Clin. Oncol. 2010, 28, 780–787. [Google Scholar] [CrossRef]
- Refolo, M.G.; Lotesoriere, C.; Lolli, I.R.; Messa, C.; D’Alessandro, R. Molecular Mechanisms of Synergistic Action of Ramucirumab and Paclitaxel in Gastric Cancers Cell Lines. Sci. Rep. 2020, 10, 7162. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Schiller, J.H.; Fruehauf, J.P.; Cohen, E.E.W.; Tarazi, J.; Rosbrook, B.; Kim, S.J.; Bair, A.H.; Macek, J.; Wood, J. Diastolic Blood Pressure as a Biomarker of Axitinib Efficacy in Solid Tumors. Clin. Cancer Res. 2011, 17, 3841–3849. [Google Scholar] [CrossRef] [PubMed]
- Rixe, O.; Bukowski, R.M.; Michaelson, M.D.; Wilding, G.; Hudes, G.R.; Bolte, O.; Motzer, R.J. Axitinib Treatment in Patients with Cytokine-Refractory Metastatic Renal-Cell Cancer: A Phase II Study. Lancet Oncol. 2007, 8, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P.; Hutson, T.E.; Michaelson, M.D.; Rixe, O.; Kaelin, W.G.; Stein, M.N.; Law, C.; Ho, P.T.; Wu, B.; Motzer, R.J. Sequential Axitinib (AG-013736) Therapy of Patients with Metastatic Clear Cell Renal Cell Cancer (RCC) Refractory to Sunitinib and Sorafenib, Cytokines and Sorafenib, or Sorafenib Alone. J. Clin. Oncol. 2008, 26, 5127. [Google Scholar] [CrossRef]
- Duerinck, J.; Du Four, S.; Bouttens, F.; Andre, C.; Verschaeve, V.; Van Fraeyenhove, F.; Chaskis, C.; D’Haene, N.; Le Mercier, M.; Rogiers, A.; et al. Randomized Phase II Trial Comparing Axitinib with the Combination of Axitinib and Lomustine in Patients with Recurrent Glioblastoma. J. Neurooncol. 2018, 136, 115–125. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.-M.; Michotte, A.; Everaert, H.; et al. Axitinib plus Avelumab in the Treatment of Recurrent Glioblastoma: A Stratified, Open-Label, Single-Center Phase 2 Clinical Trial (GliAvAx). J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef]
- Phadnis, S.; Hagiwara, M.; Yaffe, A.; Mitchell, C.; Nicolaides, T.; Akshintala, S.; Hochman, T.; Goldberg, J.; Allen, J.; Karajannis, M. NFB-08. Phase II study of axitinib in patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro Oncol. 2020, 22, iii419. [Google Scholar] [CrossRef]
- Reardon, D.A.; Vredenburgh, J.J.; Desjardins, A.; Peters, K.; Gururangan, S.; Sampson, J.H.; Marcello, J.; Herndon, J.E.; McLendon, R.E.; Janney, D.; et al. Effect of CYP3A-Inducing Anti-Epileptics on Sorafenib Exposure: Results of a Phase II Study of Sorafenib plus Daily Temozolomide in Adults with Recurrent Glioblastoma. J. Neurooncol. 2011, 101, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A.; et al. NABTT 0502: A Phase II and Pharmacokinetic Study of Erlotinib and Sorafenib for Patients with Progressive or Recurrent Glioblastoma Multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef]
- Den, R.B.; Kamrava, M.; Sheng, Z.; Werner-Wasik, M.; Dougherty, E.; Marinucchi, M.; Lawrence, Y.R.; Hegarty, S.; Hyslop, T.; Andrews, D.W.; et al. A Phase I Study of the Combination of Sorafenib with Temozolomide and Radiation Therapy for the Treatment of Primary and Recurrent High-Grade Gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Supko, J.G.; Rosenfeld, M.; Chamberlain, M.; Phuphanich, S.; Batchelor, T.; Desideri, S.; Ye, X.; Wright, J.; Gujar, S.; et al. Phase I Trial of Sorafenib in Patients with Recurrent or Progressive Malignant Glioma. Neuro Oncol. 2011, 13, 1324–1330. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Ben Aissa, A.; Espeli, V.; Squiban, D.; Dunkel, N.; Vargas, M.I.; Hundsberger, T.; Mach, N.; Schaller, K.; Weber, D.C.; et al. Phase I Study of Sorafenib Combined with Radiation Therapy and Temozolomide as First-Line Treatment of High-Grade Glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kuhn, J.; Lamborn, K.R.; Abrey, L.E.; DeAngelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.A.; Drappatz, J.; et al. Phase I/II Study of Sorafenib in Combination with Erlotinib for Recurrent Glioblastoma as Part of a 3-Arm Sequential Accrual Clinical Trial: NABTC 05-02. Neurooncol. Adv. 2020, 2, vdaa124. [Google Scholar] [CrossRef]
- Reardon, D.A.; Pan, E.; Fan, J.; Mink, J.; Barboriak, D.P.; Vredenburgh, J.J.; Desjardins, A.; Peters, K.; O’Brien, J.P.; Wen, P.Y. A Phase 2 Trial of the Multitargeted Kinase Inhibitor Lenvatinib (E7080) in Patients (PTS) with Recurrent Glioblastoma (GBM) And Disease Progression Following Prior Bevacizumab Treatment. Ann. Oncol. 2012, 23, ix146. [Google Scholar] [CrossRef]
- Lwin, Z.; Gomez-Roca, C.; Saada-Bouzid, E.; Yanez, E.; Munoz, F.L.; Im, S.A.; Castanon, E.; Senellart, H.; Graham, D.; Voss, M.; et al. LEAP-005: Phase II Study of Lenvatinib (Len) plus Pembrolizumab (Pembro) in Patients (Pts) with Previously Treated Advanced Solid Tumours. Ann. Oncol. 2020, 31, S1170. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Butowski, N.A.; Chang, S.M.; Prados, M.D.; Fine, H.A. Phase II Trial of Pazopanib (GW786034), an Oral Multi-Targeted Angiogenesis Inhibitor, for Adults with Recurrent Glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 2010, 12, 855–861. [Google Scholar] [CrossRef]
- Reardon, D.A.; Groves, M.D.; Wen, P.Y.; Nabors, L.; Mikkelsen, T.; Rosenfeld, S.; Raizer, J.; Barriuso, J.; McLendon, R.E.; Suttle, A.B.; et al. A Phase I/II Trial of Pazopanib in Combination with Lapatinib in Adult Patients with Relapsed Malignant Glioma. Clin. Cancer Res. 2013, 19, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Burzynski, S.R.; Janicki, T.J.; Burzynski, G.S.; Brookman, S. Preliminary Findings on the Use of Targeted Therapy with Pazopanib and Other Agents in Combination with Sodium Phenylbutyrate in the Treatment of Glioblastoma Multiforme. J. Cancer Ther. 2014, 5, 1423–1437. [Google Scholar] [CrossRef]
- Saada-Bouzid, E.; Frenel, J.-S.; Augereau, P.; Bourg, V.; Gal, J.; Jacquinot, F.; Gourmelon, C.; Chateau, Y.; Barriere, J.; Bondiau, P.-Y. Phase I/II Study of Pazopanib and Temozolomide in Patients with Newly Diagnosed and Resected Glioblastoma: Pazoglio Trial. J. Clin. Oncol. 2023, 41, e14024. [Google Scholar] [CrossRef]
- Wuthrick, E.J.; Kamrava, M.; Curran, W.J.; Werner-Wasik, M.; Camphausen, K.A.; Hyslop, T.; Axelrod, R.; Andrews, D.W.; Glass, J.; MacHtay, M.; et al. A Phase 1b Trial of the Combination of the Antiangiogenic Agent Sunitinib and Radiation Therapy for Patients with Primary and Metastatic Central Nervous System Malignancies. Cancer 2011, 117, 5548–5559. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Chaskis, C.; Dujardin, M.; Everaert, H.; Lv, S.; Duerinck, J.; Tynninen, O.; Nupponen, N.; Michotte, A.; et al. Phase II Study of Sunitinib Malate in Patients with Recurrent High-Grade Glioma. J. Neurooncol. 2011, 103, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Du Four, S.; Sander, W.; Van Binst, A.-M.; Everaert, H.; Michotte, A.; Hau, P.; Neyns, B. Sunitinib Malate plus Lomustine for Patients with Temozolomide-Refractory Recurrent Anaplastic or Low-Grade Glioma. Anticancer Res. 2015, 35, 5551–5557. [Google Scholar] [PubMed]
- Wetmore, C.; Daryani, V.M.; Billups, C.A.; Boyett, J.M.; Leary, S.; Tanos, R.; Goldsmith, K.C.; Stewart, C.F.; Blaney, S.M.; Gajjar, A. Phase II Evaluation of Sunitinib in the Treatment of Recurrent or Refractory High-Grade Glioma or Ependymoma in Children: A Children’s Oncology Group Study ACNS1021. Cancer Med. 2016, 5, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Wuthrick, E.J.; Curran, W.J.; Camphausen, K.; Lin, A.; Glass, J.; Evans, J.; Andrews, D.W.; Axelrod, R.; Shi, W.; Werner-Wasik, M.; et al. A Pilot Study of Hypofractionated Stereotactic Radiation Therapy and Sunitinib in Previously Irradiated Patients with Recurrent High-Grade Glioma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Faye, M.D.; Easaw, J.; De Robles, P.; Agnihotram, R.; Torres-Vasquez, A.; Lamonde, F.; Petrecca, K.; Owen, S.; Panet-Raymond, V.; Shenouda, G.; et al. Phase II Trial of Concurrent Sunitinib, Temozolomide, and Radiotherapy with Adjuvant Temozolomide for Newly Diagnosed MGMT Unmethylated Glioblastoma. Neurooncol. Adv. 2023, 5, vdad106. [Google Scholar] [CrossRef]
- Janssen, J.B.E.; Brahm, C.G.; Driessen, C.M.L.; Nuver, J.; Labots, M.; Kouwenhoven, M.C.M.; Aliaga, E.S.; Enting, R.H.; de Groot, J.C.; Walenkamp, A.M.E.; et al. The STELLAR Trial: A Phase II/III Randomized Trial of High-Dose, Intermittent Sunitinib in Patients with Recurrent Glioblastoma. Brain Commun. 2024, 6, fcae241. [Google Scholar] [CrossRef]
- Kaley, T.J.; Wen, P.; Schiff, D.; Ligon, K.; Haidar, S.; Karimi, S.; Lassman, A.B.; Nolan, C.P.; De Angelis, L.M.; Gavrilovic, I.; et al. Phase II Trial of Sunitinib for Recurrent and Progressive Atypical and Anaplastic Meningioma. Neuro Oncol. 2015, 17, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.F.; Ruiz-Patiño, A.; Zatarain-Barrón, Z.L.; Hakim, F.; Jiménez, E.; Mejía, J.A.; Ramón, J.F.; Useche, N.; Bermúdez, S.; Pineda, D.; et al. Systemic Management of Malignant Meningiomas: A Comparative Survival and Molecular Marker Analysis between Octreotide in Combination with Everolimus and Sunitinib. PLoS ONE 2019, 14, e0217340. [Google Scholar] [CrossRef]
- Schiff, D.; Desjardins, A.; Cloughesy, T.; Mikkelsen, T.; Glantz, M.; Chamberlain, M.C.; Reardon, D.A.; Wen, P.Y. Phase 1 Dose Escalation Trial of the Safety and Pharmacokinetics of Cabozantinib Concurrent with Temozolomide and Radiotherapy or Temozolomide after Radiotherapy in Newly Diagnosed Patients with High-Grade Gliomas. Cancer 2016, 122, 582–587. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Drappatz, J.; De Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Ping, J.; et al. Phase II Study of Cabozantinib in Patients with Progressive Glioblastoma: Subset Analysis of Patients with Prior Antiangiogenic Therapy. Neuro Oncol. 2018, 20, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Muhic, A.; Poulsen, H.S.; Sorensen, M.; Grunnet, K.; Lassen, U. Phase II Open-Label Study of Nintedanib in Patients with Recurrent Glioblastoma Multiforme. J. Neurooncol. 2013, 111, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Schiff, D.; Ahluwalia, M.S.; Lesser, G.J.; Nayak, L.; Lee, E.Q.; Rinne, M.L.; Muzikansky, A.; Dietrich, J.; Purow, B.; et al. Phase II Trial of Triple Tyrosine Kinase Receptor Inhibitor Nintedanib in Recurrent High-Grade Gliomas. J. Neurooncol. 2015, 121, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib Compared with Lomustine in Patients with Relapsed Glioblastoma (REGOMA): A Multicentre, Open-Label, Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Chiesa, S.; Mangraviti, A.; Martini, M.; Cenci, T.; Mazzarella, C.; Gaudino, S.; Bracci, S.; Martino, A.; Della Pepa, G.M.; Offi, M.; et al. Clinical and NGS Predictors of Response to Regorafenib in Recurrent Glioblastoma. Sci. Rep. 2022, 12, 16265. [Google Scholar] [CrossRef]
- Rudà, R.; Bruno, F.; Pellerino, A.; Pronello, E.; Palmiero, R.; Bertero, L.; Crasto, S.; Polo, V.; Vitaliani, R.; Trincia, E.; et al. Observational Real-Life Study on Regorafenib in Recurrent Glioblastoma: Does Dose Reduction Reduce Toxicity While Maintaining the Efficacy? J. Neurooncol. 2022, 160, 389–402. [Google Scholar] [CrossRef]
- Fasano, M.; Pirozzi, M.; Famiglietti, V.; Facchini, S.; Caterino, M.; Caroprese, M.; Barillaro, A.; Di Giovanni, I.; Auriemma, A.; Fattoruso, S.I.S.; et al. Clinical Activity of Regorafenib in Elderly Patients with Recurrent Glioblastoma. Mol. Clin. Oncol. 2023, 18, 9. [Google Scholar] [CrossRef]
- Nayak, L.; de Groot, J.; Wefel, J.S.; Cloughesy, T.F.; Lieberman, F.; Chang, S.M.; Omuro, A.; Drappatz, J.; Batchelor, T.T.; DeAngelis, L.M.; et al. Phase I Trial of Aflibercept (VEGF Trap) with Radiation Therapy and Concomitant and Adjuvant Temozolomide in Patients with High-Grade Gliomas. J. Neurooncol. 2017, 132, 181. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.F.; Lamborn, K.R.; Chang, S.M.; Gilbert, M.R.; Cloughesy, T.F.; Aldape, K.; Yao, J.; Jackson, E.F.; Lieberman, F.; Robins, H.I.; et al. Phase II Study of Aflibercept in Recurrent Malignant Glioma: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2011, 29, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Richly, H.; Hilger, R.A.; Schleucher, N.; Korfee, S.; Tewes, M.; Faghih, M.; Brendel, E.; Voliotis, D.; Haase, C.G.; et al. Phase I Clinical and Pharmacokinetic Study of the Novel Raf Kinase and Vascular Endothelial Growth Factor Receptor Inhibitor BAY 43-9006 in Patients with Advanced Refractory Solid Tumors. J. Clin. Oncol. 2005, 23, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Ratain, M.J.; Eisen, T.; Stadler, W.M.; Flaherty, K.T.; Kaye, S.B.; Rosner, G.L.; Gore, M.; Desai, A.A.; Patnaik, A.; Xiong, H.Q.; et al. Phase II Placebo-Controlled Randomized Discontinuation Trial of Sorafenib in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2006, 24, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lang, L. FDA Approves Sorafenib for Patients With Inoperable Liver Cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- McFarland, D.C.; Misiukiewicz, K.J. Sorafenib in Radioactive Iodine-Refractory Well-Differentiated Metastatic Thyroid Cancer. Onco Targets Ther. 2014, 7, 1291–1299. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Cascinu, S.; Berardi, R.; Sobrero, A.; Bidoli, P.; Labianca, R.; Siena, S.; Ferrari, D.; Barni, S.; Aitini, E.; Zagonel, V.; et al. Sorafenib Does Not Improve Efficacy of Chemotherapy in Advanced Pancreatic Cancer: A GISCAD Randomized Phase II Study. Dig. Liver Dis. 2014, 46, 182–186. [Google Scholar] [CrossRef]
- Ott, P.A.; Hamilton, A.; Min, C.; Safarzadeh-Amiri, S.; Goldberg, L.; Yoon, J.; Yee, H.; Buckley, M.; Christos, P.J.; Wright, J.J.; et al. A Phase II Trial of Sorafenib in Metastatic Melanoma with Tissue Correlates. PLoS ONE 2010, 5, e15588. [Google Scholar] [CrossRef]
- Chauhan, S.J.; Thyagarajan, A.; Sahu, R.P. Effects of MiRNA-149-5p and Platelet-Activating Factor-Receptor Signaling on the Growth and Targeted Therapy Response on Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 6772. [Google Scholar] [CrossRef]
- Bronte, G.; Andreis, D.; Bravaccini, S.; Maltoni, R.; Cecconetto, L.; Schirone, A.; Farolfi, A.; Fedeli, A.; Serra, P.; Donati, C.; et al. Sorafenib for the Treatment of Breast Cancer. Expert Opin. Pharmacother. 2017, 18, 621–630. [Google Scholar] [CrossRef]
- Karajannis, M.A.; Legault, G.; Fisher, M.J.; Milla, S.S.; Cohen, K.J.; Wisoff, J.H.; Harter, D.H.; Goldberg, J.D.; Hochman, T.; Merkelson, A.; et al. Phase II Study of Sorafenib in Children with Recurrent or Progressive Low-Grade Astrocytomas. Neuro Oncol. 2014, 16, 1408. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Schlumberger, M.; Jarzab, B.; Martins, R.G.; Pacini, F.; Robinson, B.; McCaffrey, J.C.; Shah, M.H.; Bodenner, D.L.; Topliss, D.; et al. A Phase 2 Trial of Lenvatinib (E7080) in Advanced, Progressive, Radioiodine-Refractory, Differentiated Thyroid Cancer: A Clinical Outcomes and Biomarker Assessment. Cancer 2015, 121, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Lemery, S.J.; Yang, J.; Marathe, A.; Zhao, L.; Zhao, H.; Jiang, X.; He, K.; Ladouceur, G.; Mitra, A.K.; et al. FDA Approval Summary: Lenvatinib for Progressive, Radio-Iodine-Refractory Differentiated Thyroid Cancer. Clin. Cancer Res. 2015, 21, 5205–5208. [Google Scholar] [CrossRef]
- Federal Drug Administration FDA Approves Drug Combo for Kidney Cancer. Cancer Discov. 2016, 6, 687–688. [CrossRef] [PubMed]
- Personeni, N.; Pressiani, T.; Rimassa, L. Lenvatinib for the Treatment of Unresectable Hepatocellular Carcinoma: Evidence to Date. J. Hepatocell. Carcinoma 2019, 6, 31–39. [Google Scholar] [CrossRef]
- Food and Drug Administration Oncology (Cancer)/Hematologic Malignancies Approval Notifications|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/oncology-cancerhematologic-malignancies-approval-notifications (accessed on 3 December 2024).
- Arance, A.; de la Cruz-Merino, L.; Petrella, T.M.; Jamal, R.; Ny, L.; Carneiro, A.; Berrocal, A.; Márquez-Rodas, I.; Spreafico, A.; Atkinson, V.; et al. Phase II LEAP-004 Study of Lenvatinib Plus Pembrolizumab for Melanoma With Confirmed Progression on a Programmed Cell Death Protein-1 or Programmed Death Ligand 1 Inhibitor Given as Monotherapy or in Combination. J. Clin. Oncol. 2023, 41, 75–85. [Google Scholar] [CrossRef]
- Shalata, W.; Iraqi, M.; Bhattacharya, B.; Fuchs, V.; Roisman, L.C.; Cohen, A.Y.; Massalha, I.; Yakobson, A.; Prasad, M.; Elkabets, M.; et al. Rapid Response to the Combination of Lenvatinib and Pembrolizumab in Patients with Advanced Carcinomas (Lung Adenocarcinoma and Malignant Pleural Mesothelioma). Cancers 2021, 13, 3630. [Google Scholar] [CrossRef]
- González-Martín, A.; Chung, H.C.; Saada-Bouzid, E.; Yanez, E.; Senellart, H.; Cassier, P.A.; Basu, B.; Corr, B.R.; Girda, E.; Dutcus, C.; et al. Lenvatinib plus Pembrolizumab for Patients with Previously Treated Advanced Ovarian Cancer: Results from the Phase 2 Multicohort LEAP-005 Study. Gynecol. Oncol. 2024, 186, 182–190. [Google Scholar] [CrossRef]
- Chung, H.C.; Saada-Bouzid, E.; Longo, F.; Yanez, E.; Im, S.-A.; Castanon, E.; Desautels, D.N.; Graham, D.M.; Garcia-Corbacho, J.; Lopez, J.; et al. Lenvatinib plus Pembrolizumab for Patients with Previously Treated, Advanced, Triple-Negative Breast Cancer: Results from the Triple-Negative Breast Cancer Cohort of the Phase 2 LEAP-005 Study. Cancer 2024, 130, 3278–3288. [Google Scholar] [CrossRef]
- Ueno, M.; Ikeda, M.; Sasaki, T.; Nagashima, F.; Mizuno, N.; Shimizu, S.; Ikezawa, H.; Hayata, N.; Nakajima, R.; Morizane, C. Phase 2 Study of Lenvatinib Monotherapy as Second-Line Treatment in Unresectable Biliary Tract Cancer: Primary Analysis Results. BMC Cancer 2020, 20, 1105. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Fazio, N.; Lopez, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Lenvatinib in Patients With Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J. Clin. Oncol. 2021, 39, 2304–2312. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- van der Graaf, W.T.A.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for Metastatic Soft-Tissue Sarcoma (PALETTE): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; Bukowski, R.M.; Curti, B.D.; George, D.J.; Hudes, G.R.; Redman, B.G.; Margolin, K.A.; Merchan, J.R.; Wilding, G.; et al. Sunitinib in Patients with Metastatic Renal Cell Carcinoma. JAMA 2006, 295, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, G.M.; Cortazar, P.; Zhang, J.J.; Tang, S.; Sridhara, R.; Murgo, A.; Justice, R.; Pazdur, R. FDA Approval Summary: Sunitinib for the Treatment of Progressive Well-Differentiated Locally Advanced or Metastatic Pancreatic Neuroendocrine Tumors. Oncologist 2012, 17, 1108–1113. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Smith, P.; Sul, J.; Salgado, C.; Iwamoto, F.M.; Shih, J.H.; Fine, H.A. Continuous Daily Sunitinib for Recurrent Glioblastoma. J. Neurooncol. 2013, 111, 41–48. [Google Scholar] [CrossRef]
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A Single-Arm Phase II Austrian/German Multicenter Trial on Continuous Daily Sunitinib in Primary Glioblastoma at First Recurrence (SURGE 01-07). Neuro Oncol. 2014, 16, 92–102. [Google Scholar] [CrossRef]
- Pan, E.; Yu, D.; Yue, B.; Potthast, L.; Chowdhary, S.; Smith, P.; Chamberlain, M. A Prospective Phase II Single-Institution Trial of Sunitinib for Recurrent Malignant Glioma. J. Neurooncol. 2012, 110, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Balaña, C.; Gil, M.J.; Perez, P.; Reynes, G.; Gallego, O.; Ribalta, T.; Capellades, J.; Gonzalez, S.; Verger, E. Sunitinib Administered Prior to Radiotherapy in Patients with Non-Resectable Glioblastoma: Results of a Phase II Study. Target. Oncol. 2014, 9, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Vredenburgh, J.J.; Coan, A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sathornsumetee, S.; Rich, J.N.; Herndon, J.E.; Friedman, H.S. Phase I Study of Sunitinib and Irinotecan for Patients with Recurrent Malignant Glioma. J. Neurooncol. 2011, 105, 621–627. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Powles, T.; Burotto, M.; Escudier, B.; Apolo, A.B.; Bourlon, M.T.; Shah, A.Y.; Suárez, C.; Porta, C.; Barrios, C.H.; Richardet, M.; et al. Nivolumab plus Cabozantinib versus Sunitinib for First-Line Treatment of Advanced Renal Cell Carcinoma: Extended Follow-up from the Phase III Randomised CheckMate 9ER Trial. ESMO Open 2024, 9, 102994. [Google Scholar] [CrossRef]
- American Cancer Society About Thyroid Cancer|Thyroid Cancer Overview|American Cancer Society. Available online: https://www.cancer.org/cancer/types/thyroid-cancer/about.html (accessed on 6 January 2025).
- Pulmonary Fibrosis Foundation (PFF) Pulmonary Fibrosis Foundation Applauds First-Ever FDA-Approved IPF Drugs. Available online: https://pulmonaryhypertensionnews.com/news/2-ipf-drugs-get-fda-approval-pulmonary-fibrosis-foundations-praise/ (accessed on 6 October 2014).
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in Patients with Progressive Fibrosing Interstitial Lung Diseases-Subgroup Analyses by Interstitial Lung Disease Diagnosis in the INBUILD Trial: A Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Werner, J.M.; Wollring, M.M.; Tscherpel, C.; Rosen, E.K.; Werr, L.; Stetter, I.; Rueß, D.; Ruge, M.I.; Brunn, A.; Al Shughri, A.; et al. Multimodal Imaging Findings in Patients with Glioblastoma with Extensive Coagulative Necrosis Related to Regorafenib. Neuro Oncol. 2023, 25, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Rodon, J.A.; Mason, W.; Beck, J.T.; Degroot, J.; Donnet, V.; Mills, D.; El-Hashimy, M.; Rosenthal, M. Phase I, Open-Label, Multicentre Study of Buparlisib in Combination with Temozolomide or with Concomitant Radiation Therapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma. ESMO Open 2020, 5, e000673. [Google Scholar] [CrossRef]
- Wen, P.Y.; Mellinghoff, I.K.; Buxton, M.B.; Cavenee, W.K.; Colman, H.; De Groot, J.F.; Ellingson, B.M.; Gordon, G.B.; Khasraw, M.; Lassman, A.B.; et al. GBM AGILE: A Global, Phase 2/3 Adaptive Platform Trial to Evaluate Multiple Regimens in Newly Diagnosed and Recurrent Glioblastoma. J. Clin. Oncol. 2021, 39, TPS2074. [Google Scholar] [CrossRef]
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus Placebo in Combination with Docetaxel and Prednisone for Treatment of Men with Metastatic Castration-Resistant Prostate Cancer (VENICE): A Phase 3, Double-Blind Randomised Trial. Lancet Oncol. 2013, 14, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Lakomy, R.; Tabernero, J.; Prausová, J.; Ruff, P.; Van Hazel, G.; Moiseyenko, V.M.; Ferry, D.R.; McKendrick, J.J.; Cutsem, E. Van Effects of Prior Bevacizumab (B) Use on Outcomes from the VELOUR Study: A Phase III Study of Aflibercept (Afl) and FOLFIRI in Patients (Pts) with Metastatic Colorectal Cancer (MCRC) after Failure of an Oxaliplatin Regimen. J. Clin. Oncol. 2012, 30, 3505. [Google Scholar] [CrossRef]
- De Groot, J.F.; Cloughesy, T.; Lieberman, F.S.; Chang, S.M.; Omuro, A.M.P.; Drappatz, J.; Batchelor, T.; DeAngelis, L.M.; Gilbert, M.R.; Yung, W.K.A.; et al. Phase I Study of Aflibercept (VEGF Trap) and Temozolomide in Newly Diagnosed, High-Grade Glioma. J. Clin. Oncol. 2011, 29, 2043. [Google Scholar] [CrossRef]
- Aprile, G.; Bonotto, M.; Ongaro, E.; Pozzo, C.; Giuliani, F. Critical Appraisal of Ramucirumab (IMC-1121B) for Cancer Treatment: From Benchside to Clinical Use. Drugs 2013, 73, 2003–2015. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus Paclitaxel versus Placebo plus Paclitaxel in Patients with Previously Treated Advanced Gastric or Gastro-Oesophageal Junction Adenocarcinoma (RAINBOW): A Double-Blind, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus Docetaxel versus Placebo plus Docetaxel for Second-Line Treatment of Stage IV Non-Small-Cell Lung Cancer after Disease Progression on Platinum-Based Therapy (REVEL): A Multicentre, Double-Blind, Randomised Phase 3 Trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus Placebo in Combination with Second-Line FOLFIRI in Patients with Metastatic Colorectal Carcinoma That Progressed during or after First-Line Therapy with Bevacizumab, Oxaliplatin, and a Fluoropyrimidine (RAISE): A Randomised, Double-Blind, Multicentre, Phase 3 Study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.H.; Park, K.; Novello, S.; Nadal, E.; et al. RELAY: Final Overall Survival for Erlotinib Plus Ramucirumab or Placebo in Untreated, EGFR-Mutated Metastatic NSCLC. J. Thorac. Oncol. 2024. [Google Scholar] [CrossRef]
- Blakeley, J.O.; Fisher, J.D.; Lieberman, F.S.; Lupo, J.; Nabors, L.B.; Crane, J.; Wen, P.Y.; Cote, A.; Peereboom, D.M.; Wen, Q.; et al. Imaging Biomarkers of Ramucirumab and Olaratumab in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 2044. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).