1. Introduction
Pulmonary tuberculosis is a zoonotic infectious disease caused by infection with the
Mycobacterium tuberculosis complex (MTB). In 2023, an estimated 400,000 cases of MDR-TB were found [
1]. The
BCG vaccine, a live attenuated strain of
Mycobacterium bovis, is administered to children for the prevention of tuberculosis clinically [
2].
M. tuberculosis is capable of infecting macrophages and inhibiting the maturation of phagolysosomes [
3], thereby ensuring long-term intracellular survival within macrophages [
4]. To survive within this hostile environment,
M. tuberculosis encounters various stressors, including but not limited to oxidative stress, reductive stress, acid stress, and nutrient deprivation. To promptly respond to these unfavorable environmental challenges,
M. tuberculosis possesses a sophisticated regulatory network, including a diverse array of transcription factors that orchestrate cellular responses to environmental cues [
5].
M. tuberculosis is subjected to a variety of external adverse stressors, including but not limited to oxidative stress, reductive stress, acid stress, and nutrient deprivation. To promptly respond to these unfavorable environmental challenges,
M. tuberculosis possesses an extensive array of transcription factors within its cells, ensuring a rapid and effective response to external stresses [
5]. Transcription factors need to interact with RNA polymerase to exert their transcriptional regulatory activity. RNA polymerase depends on sigma factors to recognize promoters and ensure transcription specificity. Activating transcription factors enhance transcription by altering promoter structure, interacting with sigma factors, or binding the RNA polymerase α-subunit. In contrast, repressive transcription factors inhibit mRNA synthesis by blocking RNA polymerase binding, modifying promoter topology, or antagonizing activators [
6]. To date, at least 35 transcription factors and 11 nucleoid-associated proteins have been identified in
M. tuberculosis [
5,
7]. These molecules are extensively involved in the bacterium’s response to external stressors. For example, c-di-GMP regulates the antioxidant capacity of Mycobacterium by activating LtmA and inhibiting HpoR [
8]. Whib7 mediates the resistance of Mycobacterium to aminoglycosides, β-lactams, macrolides, and quinolones [
9]. However, current research has not yet clarified which transcription factors regulate biofilm formation in Mycobacterium.
Biofilms are typically defined as “a sessile community derived from microorganisms, characterized by cells that irreversibly attach to a substrate or interface, or to one another, become embedded in a self-produced extracellular polymeric matrix, and exhibit altered phenotypes with respect to growth rate and gene transcription”. Under certain environmental stimuli, planktonic (free-floating) bacteria can initiate the transition from a planktonic state to a complex, surface-attached community structure [
10]. Biofilms are composed of biofilm cells and a matrix of hydrated extracellular polymeric substances (EPSs), with EPSs accounting for over 90% of the biofilm’s dry weight [
11]. Extensive in vitro studies have shown that biofilms contribute to the development of drug resistance in
Mycobacterium tuberculosis. Even rifampin-sensitive mutant strains can regain resistance to rifampin when grown within biofilms [
12]. In
M. tuberculosis infections, biofilm formation has been implicated in the establishment of persistent infections. Studies have demonstrated the presence of cellulose-dominant biofilms in the lung tissues of tuberculosis patients, contributing to the development of drug tolerance [
13]. However, the mechanism of regulating biofilm formation in
M. tuberculosis remains incompletely understood. Recent studies have highlighted the crucial role of nucleoid-associated proteins in biofilm formation. Lsr2 has been shown to mediate biofilm development induced by high concentrations of c-di-GMP through transcriptional upregulation of
hadD [
14].
Serine, a non-essential amino acid, plays a pivotal role in various cellular processes, including protein synthesis, one-carbon metabolism, and lipid biosynthesis. Serine de novo synthesis originates from glycolysis, where 3-phosphoglycerate (3-PG) is sequentially oxidized by SerA to form 3-phosphohydroxypyruvate (3-PHP), converted to 3-phosphoserine (3-PS) by SerC with glutamate deamination, and dephosphorylated by SerB to produce L-serine [
15]. Accordingly, SerC is necessary for serine metabolism and essential for mycobacterial growth and survival. A study on
M. smegmatis demonstrated that
serC expression is induced in response to various environmental stresses, including oxidative stress, nitric oxide (NO), membrane-denaturing agents, and antibiotic exposure [
16]. Isoniazid, rifampin, and H
2O
2 induce the upregulation of
serC expression in
M. smegmatis, while inhibiting
serC expression enhances the bactericidal effect of these agents and H
2O
2 on
M. smegmatis [
16].
In our previous screening, we identified a M. smegmatis strain exhibiting a smooth colony surface morphology. Subsequent sequencing revealed that the expression of the intracellular serC was suppressed in this strain. Here, we demonstrate that serC, a key enzyme in serine biosynthesis, plays a crucial role in various aspects of mycobacterial physiology, including growth, biofilm formation, and adaptation to environmental stresses. Furthermore, we elucidate the contrasting regulatory roles of a novel transcription factor NapR on serC in M. smegmatis and the BCG vaccine strains, highlighting the complex and strain-specific nature of these regulatory networks.
3. Discussion
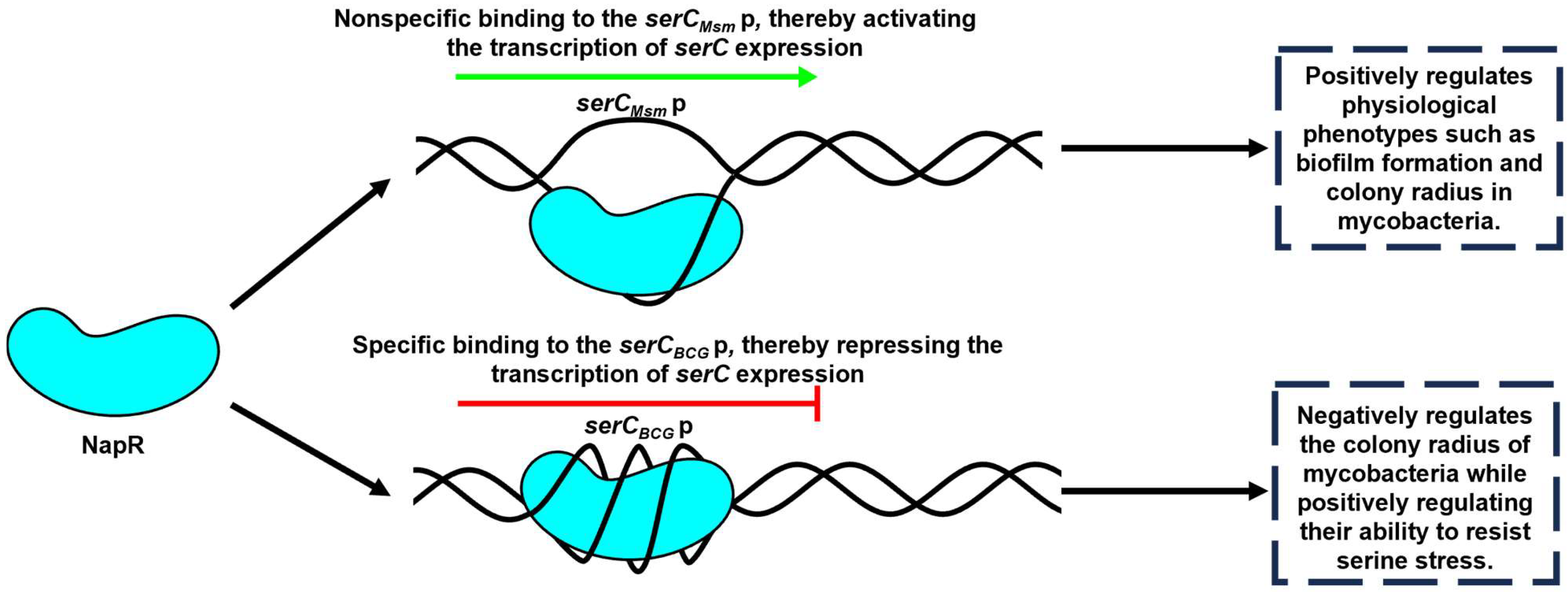
In this study, we investigated the regulatory relationship between NapR and
serC, and explored their impact on mycobacteria growth, colony morphology, and biofilm formation in both nonpathogenic strains, represented by
M. smegmatis, and pathogenic strains, represented by the
BCG vaccine. Our findings demonstrate that NapR functions as a transcriptional regulator of
serC by binding directly to the
serC promoter; however, the regulatory mechanism and its physiological consequences exhibit significant strain-specific differences. In
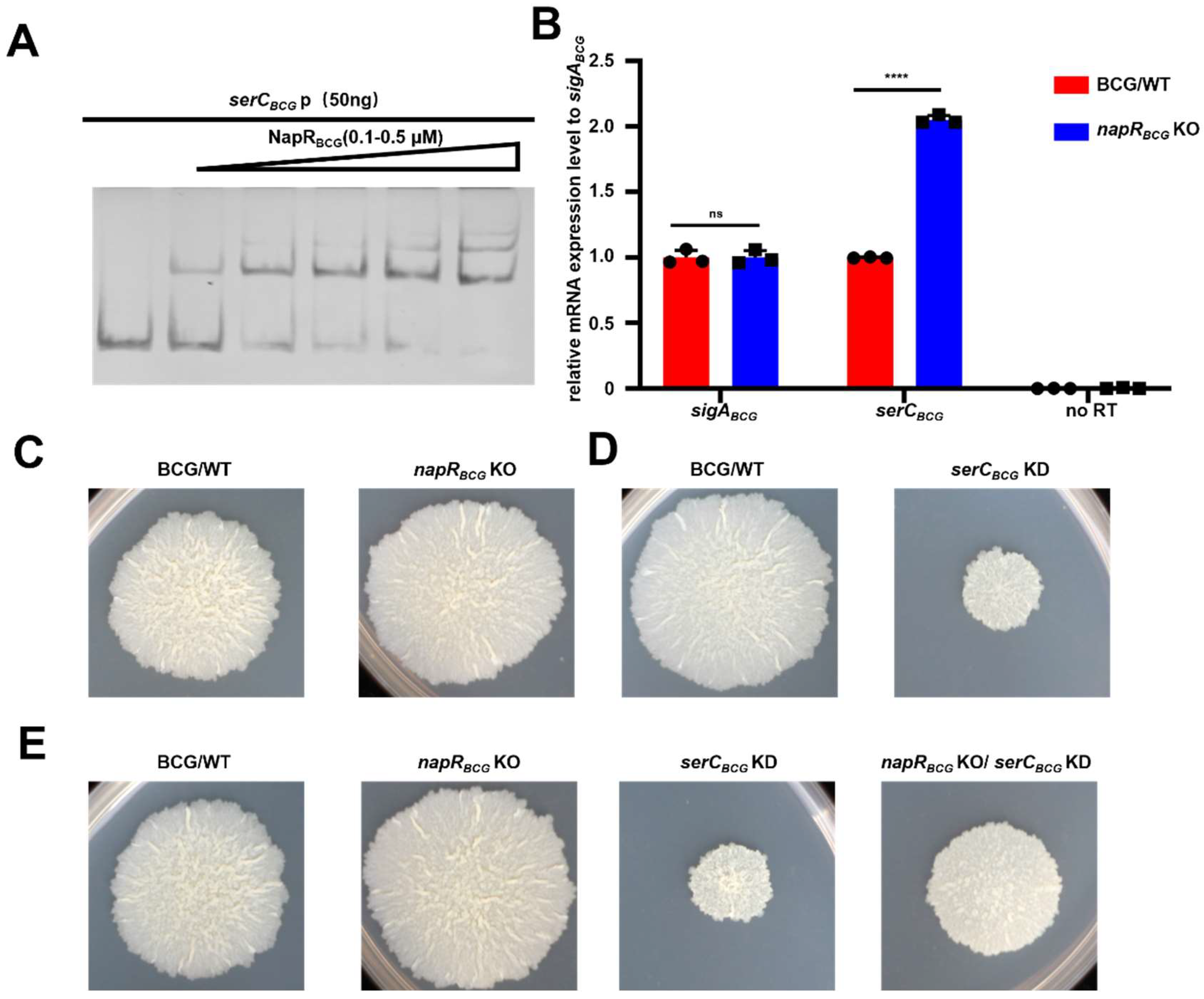
M. smegmatis, NapR
Msm positively regulates colony radius and biofilm formation by activating
serCMsm. In contrast, in the
BCG vaccine, NapR
BCG exerts a negative regulatory effect on
BCG vaccine colony size, but facilitates the
BCG vaccine’s tolerance to serine stress by repressing
serCBCG (
Figure 7).
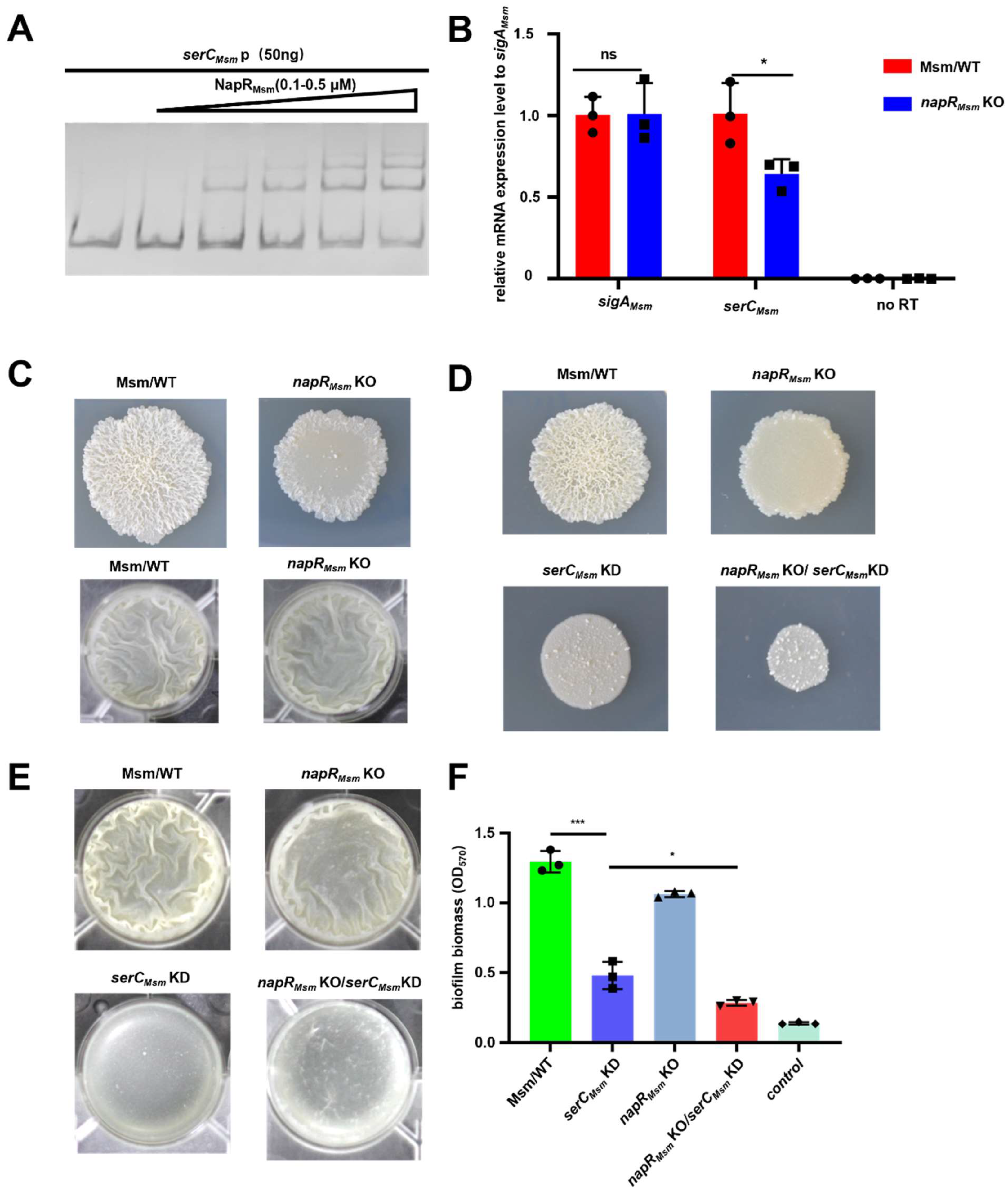
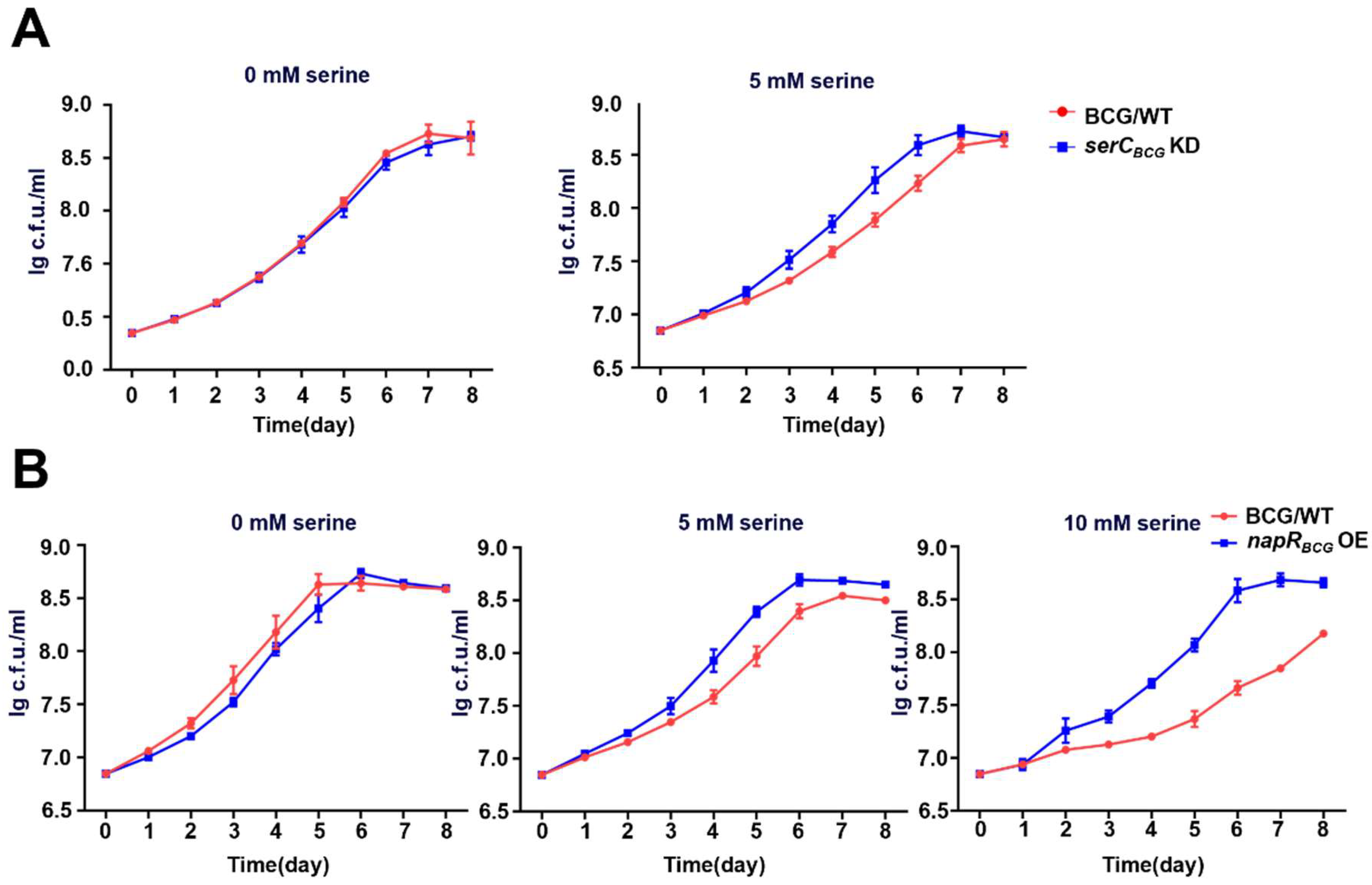
As the key enzyme in serine metabolism, SerC is essential for mycobacterial growth, with its repression leading to significant growth defects on solid media. Intriguingly, NapR activates
serC in pathogenic strains but represses
serC in nonpathogenic strains. The contrasting regulatory effects of NapR might be associated with the form and degree of its binding to the
serC promoter. EMSA experiments revealed that at a concentration of 5 μM, both NapR
Msm and NapR
BCG could fully bind 50 ng of the
serC promoter from the
BCG vaccine (
Figure 4A and
Figure S9A), whereas neither protein was unable to completely bind 50 ng of the
serC promoter from
M. smegmatis under the same conditions (
Figure 2A and
Figure S9A). Despite the high degree of homology between NapR
Msm and NapR
BCG, the sequence homology of the
serC promoter regions in these two species is relatively low (
Figure S9B). The
BCG vaccine and
M. tuberculosis share a high degree of homology. The homology between
serCBCGp and
serCMtb p is 100% (
Figure S9B), and the homology of NapR and SerC between
M. tuberculosis and
BCG vaccine is also 100% (
Figure S10B). We conclude that NapR functions as a repressor of SerC in
M. tuberculosis. Based on these findings, we speculate that variations in the
serC promoter sequence leads to the strong and specific binding of NapR to the
serC promoter in pathogenic strains, and, on the other hand, weak and nonspecific binding in nonpathogenic strains, likely contributing to the observed differences in the regulatory outcomes.
According to the widely accepted biofilm development model, biofilm formation can be summarized into reversible attachment, irreversible attachment, maturation, and dispersion [
27]. The maturation phase can be further divided into three stages: the microcolony phase, the macrocolony phase, and the mature biofilm phase [
18]. However, the precise distinctions between microcolonies and macrocolonies remain to be fully elucidated. A recent study in
Pseudomonas aeruginosa demonstrated that Psl polysaccharides are evenly distributed throughout microcolonies, whereas in macrocolonies, Psl is predominantly localized at the biofilm periphery [
28]. In our study,
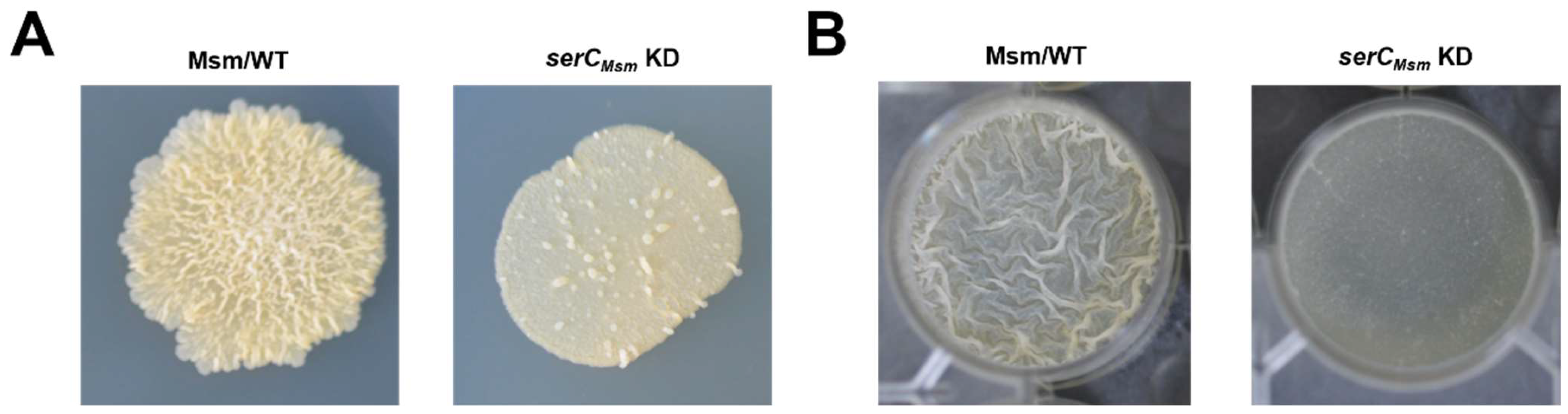
serC repression inhibited biofilm formation in
M. smegmatis (
Figure 2E,F). Further experiments showed that while
serC repression does not affect growth in static liquid cultures, plating these cultures onto 7H10 solid media not only resulted in a colony count reduced to half that of the wild-type strain, but also inhibited the transition from microcolonies to macrocolonies (
Figure 3B). Moreover, in a strain lacking
napR with concomitant
serC repression, the colony count was even reduced to approximately one-tenth that of the wild-type (
Figure 3A and
Figure S3B). This phenomenon is likely due to reduced attachment of
M. smegmatis to 7H10 agar as a result of
serC repression. The different effect in liquid and solid media can likely be attributed to nutrient availability and diffusion dynamics, as well as the cell–cell interactions and communication. Notably, this growth inhibition on solid medium could not be completely alleviated by serine supplementation, suggesting that the effects of
serC repression on solid-phase growth extend beyond serine metabolism. Although
serC is necessary for the serine biosynthesis pathway, it is also interconnected with other amino acid metabolic pathways. Therefore, serine supplementation alone could not restore the broader metabolic imbalances due to
serC repression. Altogether, our study revealed that
serC repression impairs biofilm formation in
M. smegmatis through a multi-faceted mechanism, encompassing both defects in initial attachment to solid surfaces and an inhibition of the transition from microcolonies to macrocolonies during biofilm development.
Beyond its role in biosynthesis and cell growth, serine metabolism plays a pivotal role in eukaryotic anti-infection processes. Lipopolysaccharide (LPS) can directly stimulate Toll-like receptors on macrophages, thereby activating intracellular serine synthesis [
29]. Through the provision of one-carbon units to the methionine cycle, serine promotes the production of S-adenosylmethionine (SAM), which, in turn, modifies histones or DNA via methylation and stimulates macrophages to release IL-1β [
29]. IL-1β, a key pro-inflammatory cytokine, plays a crucial role in macrophage-mediated resistance to
M. tuberculosis. It was reported that mice infected with
M. tuberculosis and lacking IL-1β exhibit significantly reduced survival rate rates compared to wild-type mice [
30]. IL-1β can also act on epithelial cells to produce antimicrobial peptides, such as DEFB4/HBD2, and promotes endothelial cell expression of adhesion molecules, facilitating the recruitment of other immune cells to sites of infection [
31]. To counteract these host defense mechanisms,
M. tuberculosis has evolved strategies to inhibit IL-1β secretion. For instance, Rv0198c, a
M. tuberculosis protein, suppresses IL-1β secretion by inhibiting inflammasome activation [
32]. As an environmental microbe frequently exposed to nutrient-poor conditions,
M. smegmatis must acquire large amounts of serine from its surroundings to survive. NapR
Msm, functioning as a transcriptional activator of
serCMsm, facilitates serine acquisition in
M. smegmatis. In contrast,
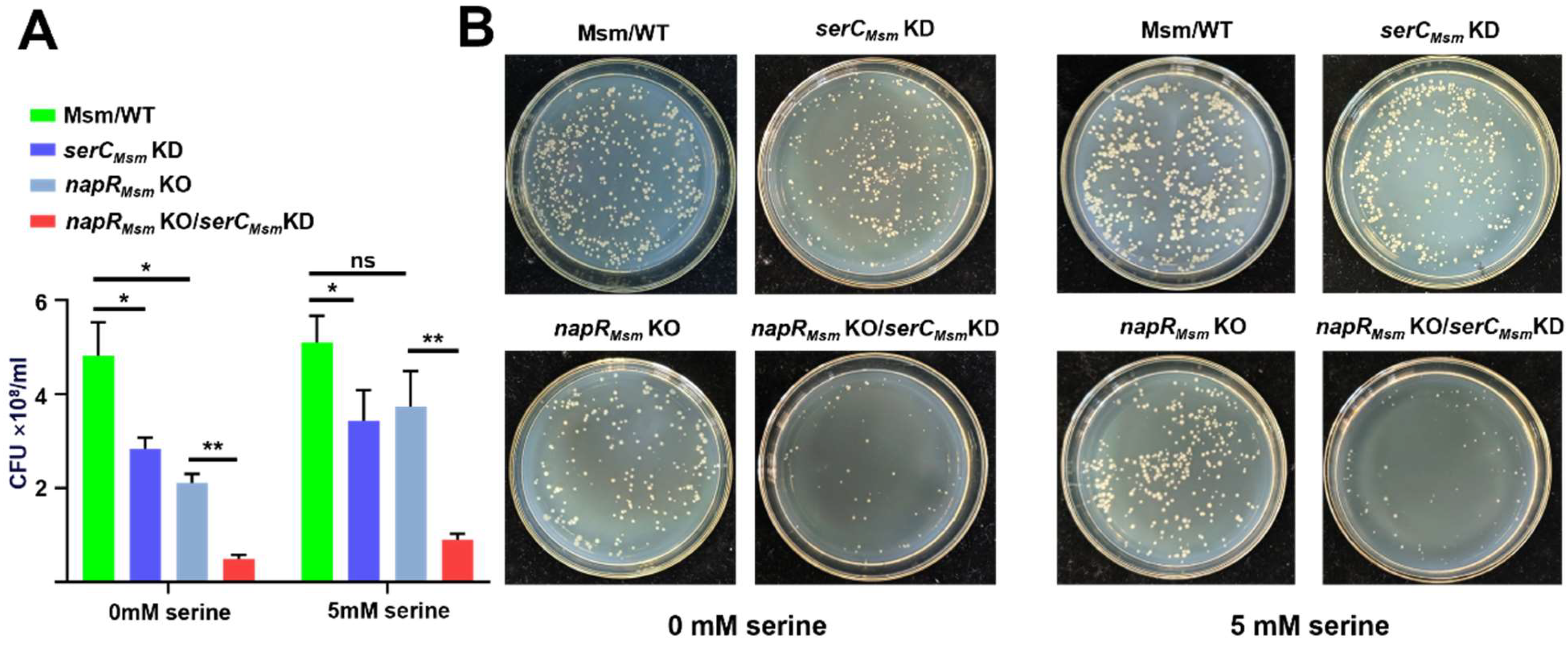
M. bovis, a pathogenic bacterium that resides within macrophages, faces a different challenge. Within the macrophage environment, high serine levels may serve as an indicator of host immune activation, particularly IL-1β production. To counteract this potential threat,
M. bovis may detect high serine levels and quickly shift into a metabolically repressed state to cope with the threat posed by macrophage activation. This inference could explain our findings that high concentrations of serine inhibit the growth of the
BCG vaccine strain, a close relative of
M. bovis, while exhibiting minimal impact on
M. smegmatis growth (
Figure S3A and
Figure 5B), suggesting that the
BCG vaccine has evolved a heightened sensitivity to serine-induced stress. Accordingly, overexpression of NapR
BCG, by repressing
serCBCG and orchestrating a coordinated modulation of other distinct serine metabolic pathways, results in a reduction in intracellular serine levels and then mitigates the detrimental effects of elevated serine levels within the macrophage environment. This mechanism enables the
BCG vaccine strain to better withstand the host immune response (
Figure 5B and
Figure 6D).
Overall, our findings demonstrate that SerC is critical for mycobacteria, and its repression has profound impacts on
M. smegmatis. Previous studies have successfully resolved the protein structure of SerC in
M. tuberculosis, revealing only 23.6% homology with human phosphoserine aminotransferase. This significant structural divergence indicates that SerC could serve as a promising new drug target for tuberculosis treatments amenable to drug discovery efforts utilizing virtual screening and molecular dynamics simulations [
33,
34]. Our findings also highlight the contrasting regulatory effects of NapR on
serC expression in pathogenic and nonpathogenic mycobacteria, suggesting a key role for NapR during human infection. Although
napR knockout in the
BCG vaccine did not produce dramatic phenotypic changes under in vitro conditions, the extensive involvement of NapR in in serine metabolism and multiple stress responses in the
BCG vaccine implies that targeting NapR may offer a novel therapeutic strategy to combat drug-resistant
M. tuberculosis and enhance host-mediated immune responses.
In conclusion, we showed that NapR functions as a transcriptional regulator of serC by binding directly to the serC promoter. In M. smegmatis, NapR activates serCMsm transcription, positively impacting biofilm formation, the transition from microcolonies to macrocolonies, and growth on solid media. In contrast, in the BCG vaccine strain, NapR functions as a transcriptional repressor of serCBCG, contributing to the BCG vaccine’s tolerance to serine stress. These findings highlight the contrasting regulatory roles of NapR in different mycobacterial species and underscore the importance of serine metabolism in bacterial adaptation and survival. This study provides valuable insights into the complex interplay between metabolism, gene regulation, and bacterial physiology, with potential implications for the development of novel anti-mycobacterial therapies.
4. Material and Methods
4.1. Cloning
Primers containing restriction sites were designed by selecting 20 bp sequences upstream and downstream of the target gene. The primers were synthesized by Tsingke Biotech (Beijing, China). The plasmid pMV261 was used to overexpress the target gene, and pET-cm1 was used for protein purification. PCR-amplified DNA fragments were ligated into the plasmids using T4 DNA ligase, and the ligated products were then chemically transformed into E. coli DH5α. The transformants were cultured in LB medium (20 g/L tryptone (OXOID, Basingstoke, UK), 10 g/L yeast extract (OXOID, Basingstoke, UK), 20 g/L NaCl (Sangon Biotech, Shanghai, China), pH 7.0), and the recombinant plasmids were extracted using the alkaline lysis method. To confirm successful construction, PCR amplification of the target gene from the recombinant plasmid was performed, and the size of the recombinant plasmid was compared with that of the original plasmid.
4.2. Protein Expression and Purification
The constructed pET-cm1 plasmid harboring the target gene was chemically transformed into E. coli BL21(DE3), followed by expansion in LB medium. The culture was incubated at 37 °C and 160 rpm until the OD600 reached 0.6. The temperature was then lowered to 16 °C, and isopropyl-β-D-1-thiogalactopyranoside (IPTG) (Solarbio, Beijing, China) was added to a final concentration of 0.4 mM. After 12 h of induction, cells were harvested by centrifugation, and the cell pellets were disrupted using a 300 W ultrasonic homogenizer. Cellular debris was collected by centrifugation at 8000× g rpm for 30 min. Ni-NTA (Solarbio, Beijing, China) agarose affinity chromatography was employed to purify proteins containing His tags: column washes were performed with 50 mM and 100 mM imidazole (Solarbio, Beijing, China), while the target protein was eluted using 250 mM imidazole. The eluted proteins were dialyzed against protein dialysis buffer (20 mM Tris-base (Sigma-Aldrich, Saint Louis, MO, USA), 100 mM NaCl, 10% v/v glycerol (Sangon Biotech, Shanghai, China)). Purified proteins were examined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and their concentrations were measured with a NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA) instrument.
4.3. Electrophoretic Mobility Shift Assay
The promoter fragment of serC was obtained via PCR. An 8% native polyacrylamide (Solarbio, Beijing, China) gel was prepared. The promoter fragment was mixed with protein dialysis buffer to form a total volume of 17.5 μL containing various concentrations of protein, along with an additional 1.5 μL of H buffer (Takara, Dalian, China) and 1 μL of 50% (v/v) glycerol. The mixture was incubated for 15 min. Afterward, the incubated solution was loaded onto the previously prepared 8% polyacrylamide gel and subjected to electrophoresis at 150 V for 1 h in TBE buffer (5.4 g/L Tris-base, 2.75 g/L boric acid (Tianjin Aopusheng Chemical Co., Ltd., Tianjin, China), 0.372 g/L Na2EDTA·2H2O (Solarbio, Beijing, China), pH 8.3). The gel was then stained with ethidium bromide (EB) (Solarbio, Beijing, China), and images were captured using a GelDoc scanner (BIO-RAD, Hercules, CA, USA).
4.4. Construction of Target Gene Knockdown/Overexpression Strains
Knockdown plasmid construction: To knock down target gene expression, potential PAM sites on the full-length serC gene were identified, and complementary paired sgRNA primers targeting serC were designed. Bsm BI restriction sites were incorporated into the primers. After annealing, the double-stranded DNA was inserted into either the pLJR 962 vector (for M. smegmatis) or the pLJR 965 vector (for the BCG vaccine) through restriction digestion and ligation, thereby creating the corresponding recombinant plasmids.
Overexpression plasmid construction: For overexpression, napR was cloned into the pMV261+ vector to generate the recombinant plasmid. A total of 200 ng of the recombinant plasmid was introduced into competent M. smegmatis or BCG vaccine cells. Electroporation was performed at 2500 V, after which the cells were allowed to recover in 7H9 liquid medium (4.9 g/L Middlebrook 7H9 Broth (BD Difco, Franklin Lakes, NJ, USA), 0.2% v/v glycerol, 0.05% v/v Tween-80). The M. smegmatis cultures were recovered for 4 h, and the BCG vaccine cultures were recovered for 24 h. The recovered cells were then plated on 7H10 solid medium (19 g/L Middlebrook 7H10 Powder (BD Difco, Franklin Lakes, NJ, USA), 0.2% v/v glycerol, 0.03 g/L kanamycin (Solarbio, Beijing, China)) and incubated—3–4 days for M. smegmatis and 25–30 days for the BCG vaccine. Primers designed to validate the presence of the recombinant plasmid were used in PCR with bacterial lysates (prepared by heating to 100 °C for 10 min), and the resulting amplicons were analyzed to confirm the successful construction of the recombinant strains.
4.5. Real-Time Quantitative PCR (RT-qPCR)
When the constructed M. smegmatis/BCG vaccine cultures reached an OD600 of 1.0, the cells were harvested and washed twice with PBS. The cell pellets were then resuspended in 200 μL of 3 mg/mL lysozyme (Solarbio, Beijing, China) and incubated at 37 °C for 30 min. Total bacterial RNA was extracted using an RNA extraction kit from Aidlab (Beijing, China). A 20 μL aliquot of the extracted RNA was treated with 1 μL of DNase I (Takara, Dalian, China) and 2 μL of 10× DNase I buffer (Takara, Dalian, China) at 37 °C for 30 min to remove any residual genomic DNA. The reaction mixture was then heated at 65 °C for 10 min to inactivate DNase I. Using a reverse transcription kit, mRNA was reverse-transcribed. The concentration of total RNA was measured with a NanoDrop, and the reaction volume for reverse transcription was set to 40 μL, which included 300 ng RNA, 2 ng random primer (Aidlab, Beijing, China), and 8 ng 5× TURE R Mix (Aidlab, Beijing, China).
Primers for qPCR were designed using sigA as the internal reference gene, where msmeg_2758 served as sigA for M. smegmatis, and bcg_2716 served as sigA for the BCG vaccine. Real-time quantitative PCR was performed using SYBR qPCR Green Master Mix (Aidlab, Beijing, China). The relative expression of the target gene was calculated as 2−ΔΔCt.
4.6. Observation of Colony Surface Morphology
Activated mycobacterial cultures were grown in 7H9 medium containing kanamycin until OD600 reached 1.0. Subsequently, 7H10 agar plates with kanamycin—and, where applicable, 0.1 ng/mL anhydrotetracycline (Solarbio, Beijing, China)—were prepared. Two microliters of the OD600 = 1.0 culture were spotted onto the surface of 7H10 agar plates (with or without anhydrotetracycline). M. smegmatis plates were incubated statically at 37 °C for 3–5 days, while BCG vaccine plates were incubated at 37 °C for 20–30 days. Colony size and surface wrinkling were then observed.
4.7. Biofilm Assays
Activated mycobacterial cultures were grown in 7H9 medium containing kanamycin to OD600 = 1.0. The cells were harvested and resuspended in M63 salt medium (13.6 g/L KH2PO4 (Solarbio, Beijing, China), 4.2 g/L KOH (Tianjin Beichen Fangzheng Reagent Factory, Tianjin, China), 1.98 g/L (NH4)2SO4 (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China), 1.08 mg/L FeSO4 (Tianjin New Technology Industrial Park Kemao Chemical Reagent Co., Ltd. Tianjin, China), 50 mL 40% (v/v) glucose (Tianjin AoboKai Chemical Co., Ltd., Tianjin, China), 10 mL 20% (v/v) casein hydrolysate (Macklin, Shanghai, China), 10 mL 100 mM MgSO4 (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China), 7 mL 100 mM CaCl2 (Tianjin Aopusheng Chemical Co., Ltd., Tianjin, China), 0.03 g/L kanamycin). The resuspended cells were then diluted in M63 medium to OD600 = 0.3, and the resulting cell suspension was divided into two portions—one supplemented with anhydrotetracycline to a final concentration of 0.1 ng/mL, and the other without anhydrotetracycline. Three milliliters of each suspension were dispensed into the wells of a 12-well plate.
Separately, the OD600 = 0.3 suspension was further diluted to OD600 = 0.1, divided into two portions (with or without anhydrotetracycline), and 100 μL of each was dispensed into the wells of a 96-well plate.
Air–Liquid Interface Biofilm Assay: The 12-well plate was incubated statically at 30 °C for 3–5 days. Biofilm formation was monitored daily, and images were taken to record changes.
Biofilm Quantification Assay: The 96-well plate was incubated at 37 °C and 80 rpm for 36–48 h. Biofilms were then stained with 120 μL of 1% crystal violet (Solarbio, Beijing, China) solution for 30 min, followed by rinsing with deionized water to remove excess dye. The remaining crystal violet bound to the biofilm was dissolved in an ethanol–acetone solution (20% (v/v) ethanol (Sangon Biotech, Shanghai, China), 80% (v/v) acetone (Sigma-Aldrich, Saint Louis, MO, USA)) for 5 min, and the absorbance at 570 nm (OD570) was measured with a microplate reader.
4.8. Growth Curve Assays
Activated mycobacterial cultures were grown in 7H9 medium containing kanamycin to OD600 = 1.0. The cells were collected, resuspended in fresh 7H9 medium, and transferred to 100 mL 7H9 medium containing kanamycin at a starting OD600 = 0.15. If CRISPRi strains were used, anhydrotetracycline was added to a final concentration of 0.1 ng/mL. For M. smegmatis, samples (2 mL) were taken every 4 h to measure OD600; for the BCG vaccine, samples were collected every 24 h. The measured OD600 values were subsequently converted to CFU counts.
4.9. Colony Counting Assays
Activated mycobacterial cultures were grown in 7H9 medium containing kanamycin to OD600 = 1.0. The cells were then harvested and resuspended in fresh 7H9 medium. The resuspended cells were inoculated into 100 mL of 7H9 medium containing kanamycin and 0.1 ng/mL anhydrotetracycline, with a starting OD600 = 0.6. Cultures were incubated statically at 37 °C for 48 h, and OD600 was recorded. The culture was then diluted 1:20,000, and 100 μL of the diluted suspension was plated onto 7H10 agar containing kanamycin. Plates were incubated statically at 37 °C for 3–5 days (for M. smegmatis) or 20–30 days (for the BCG vaccine), after which the number of colonies on the 7H10 plates were counted.
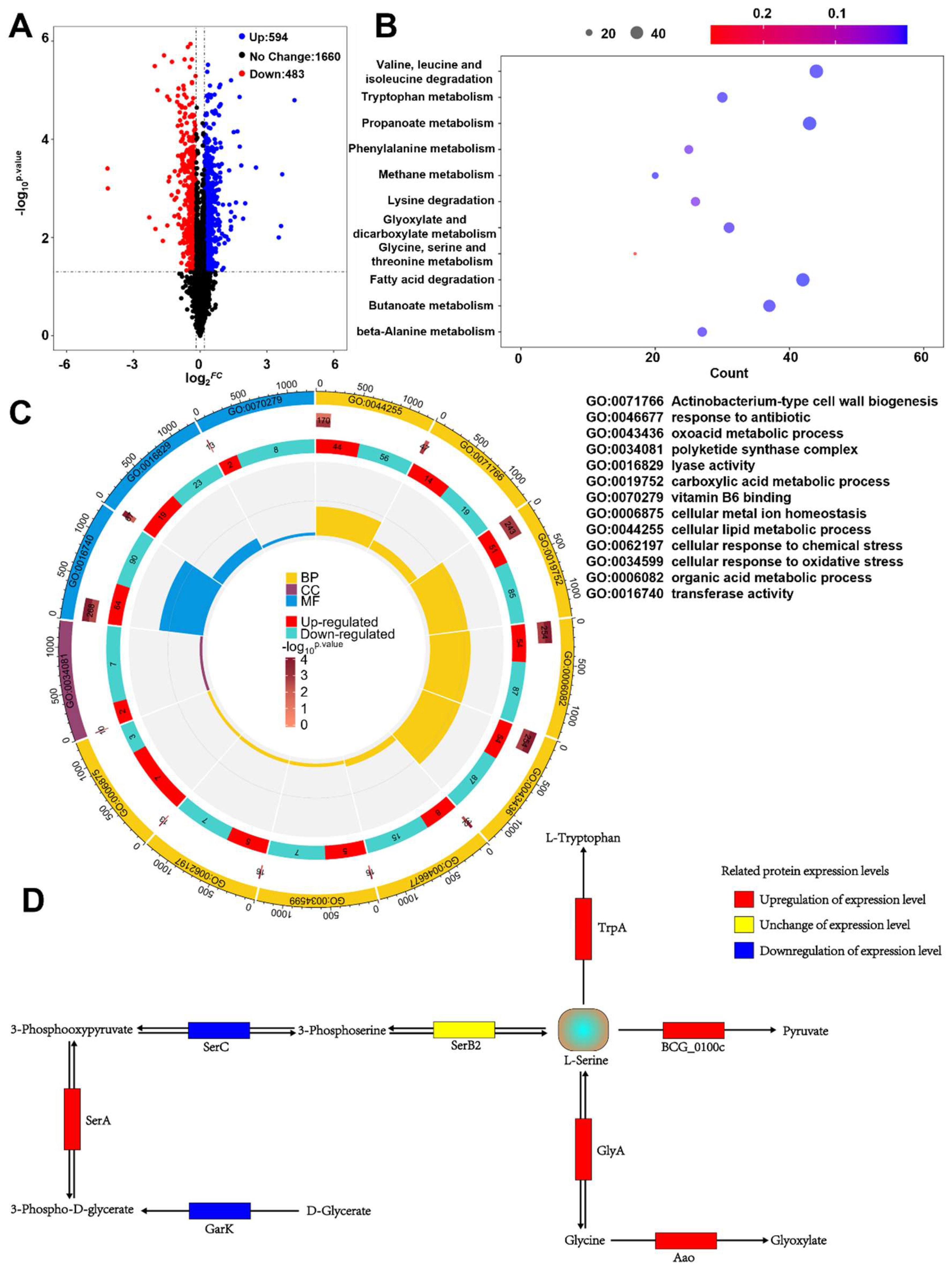
4.10. Proteomic Analysis
Activated BCG vaccine cultures were grown in 7H9 medium containing kanamycin until the OD600 reached 1.2. The cells were harvested, washed with PBS, and frozen in liquid nitrogen. Protein extraction, enzymatic digestion, and liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis were performed by Hangzhou Jingjie Biotechnology Co., Ltd. (Hangzhou, China). The bacterial pellets were then lysed in a buffer consisting of 8 M urea (Sigma-Aldrich, Saint Louis, MO, USA) and 1% protease (Merck Millipore, Billerica, MA, USA) inhibitor cocktail using a high-intensity ultrasonic processor (Scientz, Ningbo, China), followed by centrifugation to remove cell debris. Next, the proteins were reduced with 5 mM dithiothreitol (DTT) (Sigma-Aldrich, Saint Louis, MO, USA) at 56 °C for 30 min, then alkylated with 11 mM iodoacetamide (Sigma-Aldrich, Saint Louis, MO, USA) in the dark at room temperature for 15 min. The protein sample was diluted with 100 mM TEAB (Sigma-Aldrich, Saint Louis, MO, USA) until the urea concentration was below 2 M. Finally, trypsin (Promega, Madison, WI, USA) digestion was performed overnight at a trypsin-to-protein mass ratio of 1:50, followed by a second digestion for 4 h at 1:100. The resulting peptides were desalted using a C18 SPE column (ThermoFisher Scientific, Waltham, MA, USA).
After being dissolved in Liquid Phase A (0.1% formic acid (Sigma-Aldrich, Saint Louis, MO, USA), 2% acetonitrile (Thermo Fisher Scientific, Waltham, MA, USA), 98% deionized water), the peptides were separated on a NanoElute ultra-high-performance liquid chromatography (UHPLC) system (Bruker Corporation, Billerica, MA, USA). Mobile Phase A was 0.1% formic acid, 2% acetonitrile, and 98% deionized water; Mobile Phase B was 0.1% formic acid in 100% acetonitrile. The gradient was set as follows: 0–40 min, 6–24% B; 40–52 min, 24–35% B; 52–56 min, 35–80% B; 56–60 min, 80% B, at a flow rate of 450 nL/min. The separated peptides were then introduced into a capillary ion source for electrospray ionization and analyzed using a timsTOF Pro 2 mass spectrometer (Bruker Corporation, Billerica, MA, USA). The ion source voltage was set at 1.65 kV, and both the precursor ions and their corresponding fragments were detected at high resolution by the TOF analyzer. The MS/MS scan range was 100–1700 m/z. Data were acquired in parallel accumulation–serial fragmentation (PASEF) mode: after each full MS scan, 10 PASEF MS/MS scans were triggered for precursor ions with charges ranging from 0 to 5. The dynamic exclusion time was set to 30 s to avoid repeated scans of the same precursor ions.
The raw mass spectrometry data were imported into a database search engine, with the relevant analytical parameters set according to the experimental design. In this experiment, the MS2 data were processed using MaxQuant (v1.6.15.0) against the Blast_Mycobacterium_bovis_strain_BCG_410289_PR_20220526.fasta database. A reverse database was added to calculate the false discovery rate (FDR) caused by random matches, and a common contaminant database was included to eliminate the influence of contamination. The enzyme specificity was set to Trypsin/P, with up to two missed cleavage sites allowed. The minimum peptide length was set to seven amino acid residues, and a maximum of five modifications per peptide was allowed. The mass tolerances for both the first (First search) and main (Main search) searches were 20 ppm, and the MS/MS tolerance was also 20 ppm. Carbamidomethylation of cysteine (C) was set as a fixed modification, while methionine oxidation and protein N-terminal acetylation were set as variable modifications. The FDR for both protein and PSM identifications was set to 1%.
GO and KEGG enrichment analyses of the proteomic data were performed using the R language (v4.2.2) package “clusterProfiler” (v4.6.2). Volcano plots and KEGG enrichment diagrams were generated using the R package “ggplot2” (v3.5.0), while GO enrichment results were visualized with “circlize” (v0.4.16), and heatmaps were plotted using “pheatmap” (v1.0.12). Protein–protein interaction analyses were conducted via the STRING database, and protein interaction networks were visualized with the R package “igraph” (v1.4.1).
4.11. Statistical Analysis
All data are presented as the mean ± S.E.M. and represent results from at least three independent experiments. Two-tailed Student’s t-tests with equal variance assumptions were performed using Microsoft Excel (v2408, Build 16.0.17932.20252), and p-value < 0.05 was considered statistically significant.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}