
Novel Cell Models to Study Myelin and Microglia Interactions
 , , , and
, , , and
Abstract
1. Introduction
2. Results
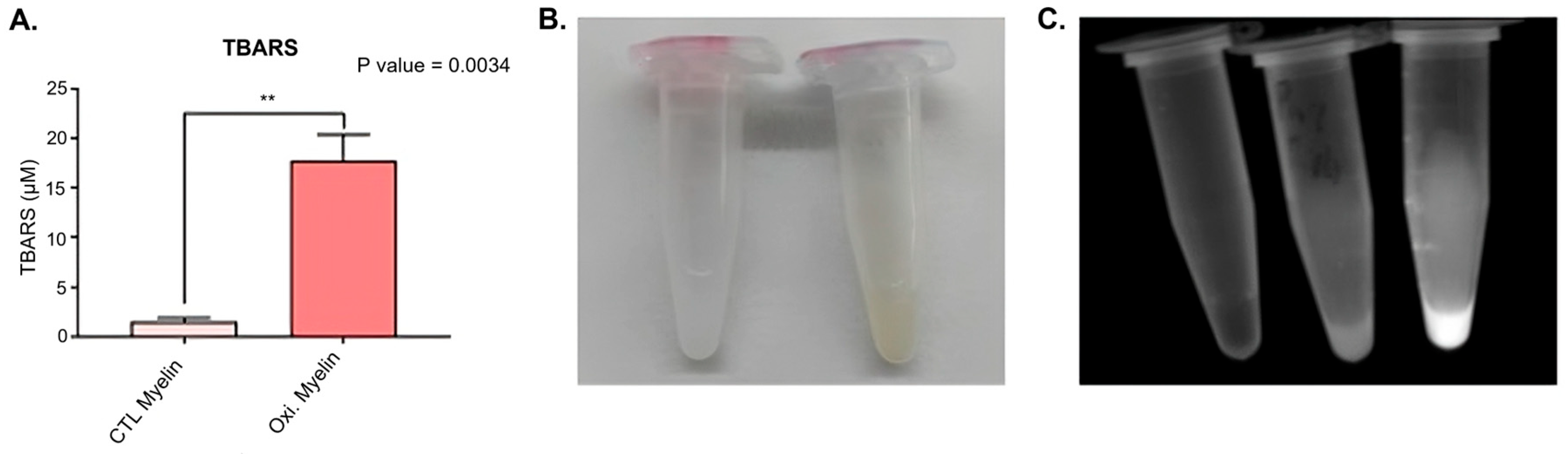
2.1. Myelin Oxidation
2.2. Cellular Viability Is Not Compromised After Myelin Exposure
2.3. Myelin Exposure Triggers Changes in Antioxidant Enzymes, Autophagy Markers, and Proteins Regulating Iron Metabolism in BV-2 Cell Line
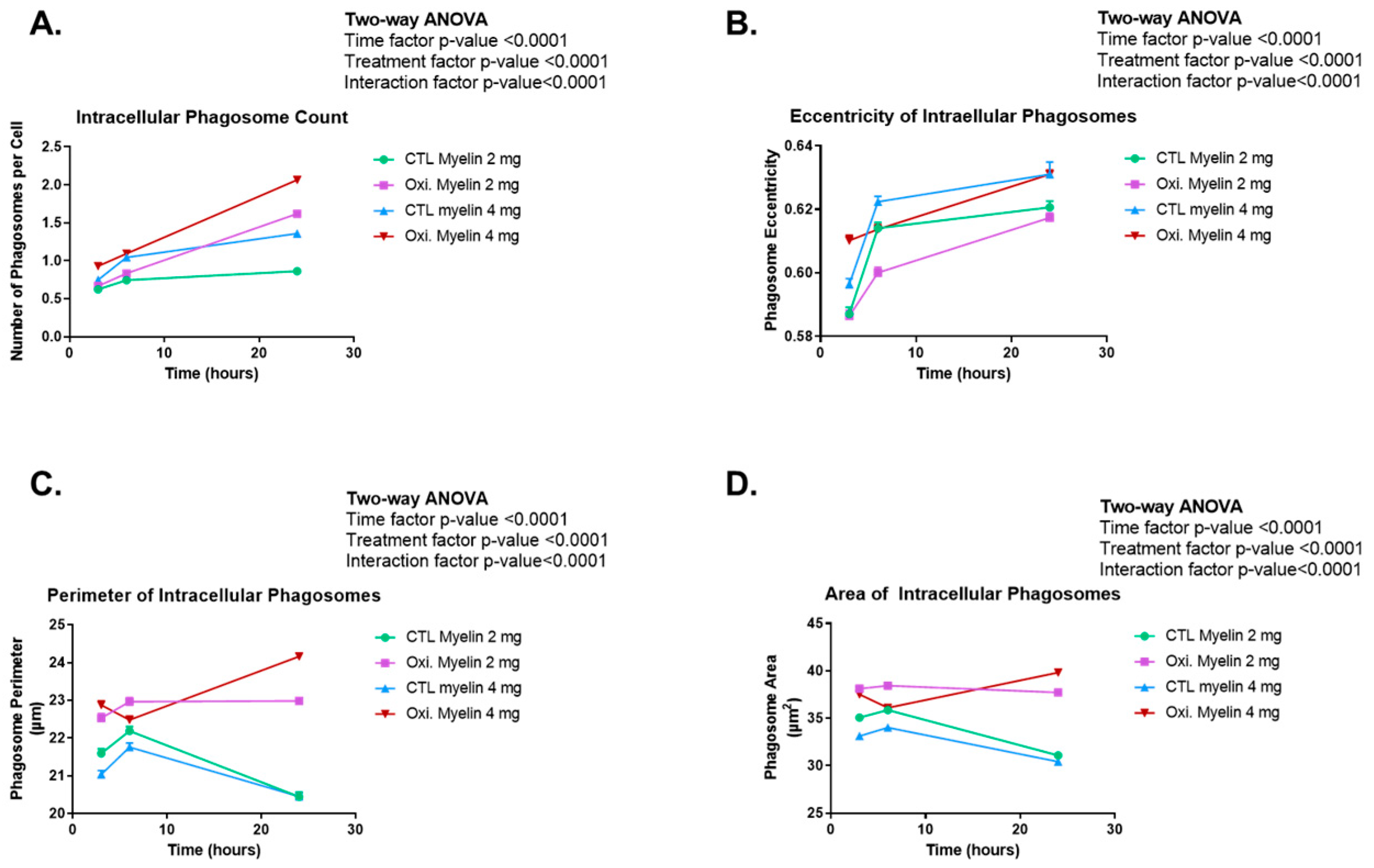
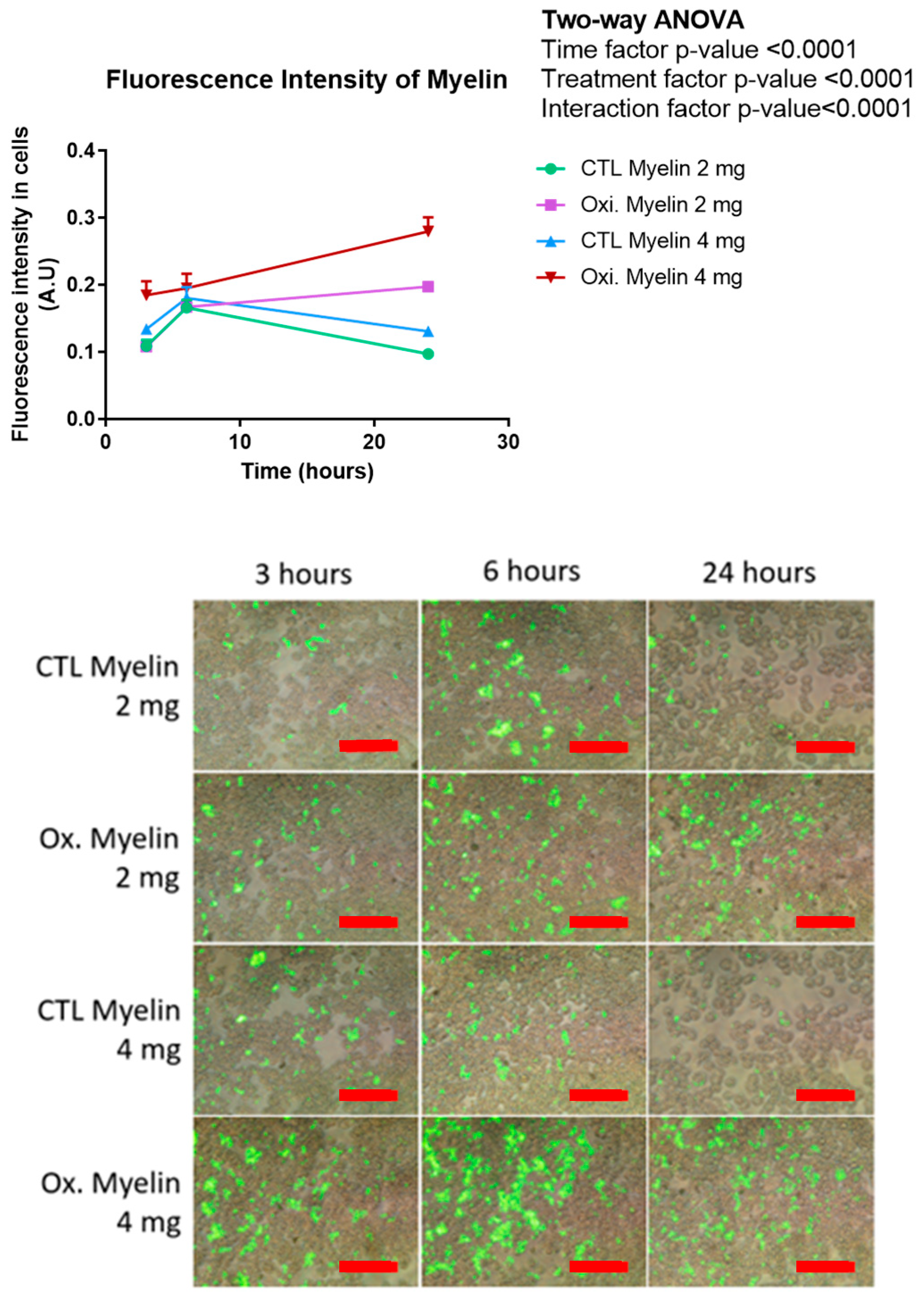
2.4. Time-Course of Morphological Analyses of Phagocyted Myelin in BV-2 Cells
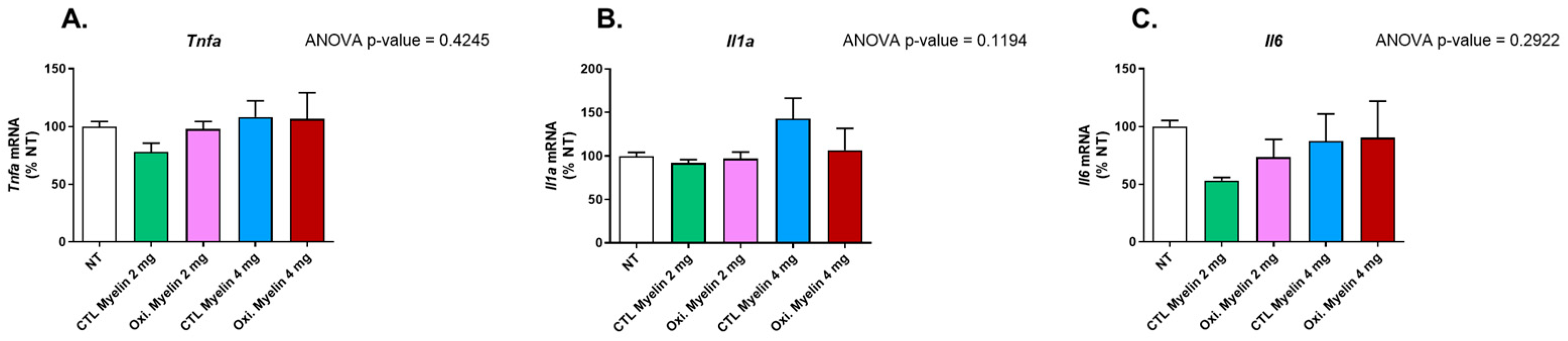
2.5. Myelin Exposure Does Not Induce a Pro-Inflammatory Profile in BV-2 Cells
2.6. Transdifferentiated Monocytes to iMG Phagocytes Myelin
3. Discussion
3.1. Oxidative Modification of Myelin
3.2. Cellular Viability and Inflammatory Profile
3.3. Oxidized Myelin and Cellular Pathways
3.4. Enhanced Phagocytosis of Oxidized Myelin
3.5. Insights from the iMG Model
3.6. Implications and Future Directions
4. Materials and Methods
4.1. Extraction and Oxidation of Myelin Debris
4.2. Labeling of Myelin Debris
4.3. Cell Cultures and Administration of Treaments
4.4. Viability Assay
4.5. Protein Expression
4.6. Study of Pro-Inflammatory Cytokines
4.7. CFSE Assay
4.8. TBARS Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MS | Multiple sclerosis |
| Oxi. | Oxidized |
| MDA | Malondialdehyde |
| TBA | Thiobarbituric acid |
| TBARS | Thiobarbituric acid reactive substances |
| CFSE | Carboxyfluorescein succinimidyl ester |
| iMG | Induced microglia-like cells |
| PBMCs | Peripheral blood mononuclear cell |
| ROS | Reactive oxygen species |
References
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive Multiple Sclerosis: From Pathophysiology to Therapeutic Strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple Sclerosis: Pathology, Diagnosis and Treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef]
- Jakimovski, D.; Bittner, S.; Zivadinov, R.; Morrow, S.A.; Benedict, R.H.; Zipp, F.; Weinstock-Guttman, B. Multiple Sclerosis. Lancet 2024, 403, 183–202. [Google Scholar] [CrossRef]
- Dighriri, I.M.; Aldalbahi, A.A.; Albeladi, F.; Tahiri, A.A.; Kinani, E.M.; Almohsen, R.A.; Alamoudi, N.H.; Alanazi, A.A.; Alkhamshi, S.J.; Althomali, N.A.; et al. An Overview of the History, Pathophysiology, and Pharmacological Interventions of Multiple Sclerosis. Cureus 2023, 15, e33242. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the Clinical Course of Multiple Sclerosis: The 2013 Revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Tur, C.; Rocca, M.A. Progression Independent of Relapse Activity in Multiple Sclerosis: Closer to Solving the Pathologic Puzzle. Neurology 2024, 102, E207936. [Google Scholar] [CrossRef]
- Husseini, L.; Geladaris, A.; Weber, M.S. Toward Identifying Key Mechanisms of Progression in Multiple Sclerosis. Trends Neurosci. 2024, 47, 58–70. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, A.M.R.; Hümmert, S.; Steyer, A.; Ruhwedel, T.; Hamann, J.; Smolders, J.; Nave, K.A.; Stadelmann, C.; Kole, M.H.P.; Möbius, W.; et al. Ultrastructural Axon-Myelin Unit Alterations in Multiple Sclerosis Correlate with Inflammation. Ann. Neurol. 2023, 93, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Mado, H.; Adamczyk-Sowa, M.; Sowa, P. Role of Microglial Cells in the Pathophysiology of MS: Synergistic or Antagonistic? Int. J. Mol. Sci. 2023, 24, 1861. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D. Progressive Multiple Sclerosis. Continuum 2019, 25, 736–752. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple Sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Murúa, S.R.; Farez, M.F.; Quintana, F.J. The Immune Response in Multiple Sclerosis. Annu. Rev. Pathol. 2022, 17, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Immunology of Relapse and Remission in Multiple Sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- van Langelaar, J.; Rijvers, L.; Smolders, J.; van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Gironi, M.; Bianchi, A.; Russo, A.; Alberoni, M.; Ceresa, L.; Angelini, A.; Cursano, C.; Mariani, E.; Nemni, R.; Kullmann, C.; et al. Oxidative Imbalance in Different Neurodegenerative Diseases with Memory Impairment. Neurodegener. Dis. 2011, 8, 129–137. [Google Scholar] [CrossRef]
- Greilberger, J.; Koidl, C.; Greilberger, M.; Lamprecht, M.; Schroecksnadel, K.; Leblhuber, F.; Fuchs, D.; Oettl, K. Malondialdehyde, Carbonyl Proteins and Albumin-Disulphide as Useful Oxidative Markers in Mild Cognitive Impairment and Alzheimer’s Disease. Free Radic. Res. 2008, 42, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Gui, L.N.; Liu, Y.Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’H, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Baldacchino, K.; Peveler, W.J.; Lemgruber, L.; Smith, R.S.; Scharler, C.; Hayden, L.; Komarek, L.; Lindsay, S.L.; Barnett, S.C.; Edgar, J.M.; et al. Myelinated Axons Are the Primary Target of Hemin-Mediated Oxidative Damage in a Model of the Central Nervous System. Exp. Neurol. 2022, 354, 114113. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. Oxidative Stress and Its Impact on Neurons and Glia in Multiple Sclerosis Lesions. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Van San, E.; Debruyne, A.C.; Veeckmans, G.; Tyurina, Y.Y.; Tyurin, V.A.; Zheng, H.; Choi, S.M.; Augustyns, K.; van Loo, G.; Michalke, B.; et al. Ferroptosis Contributes to Multiple Sclerosis and Its Pharmacological Targeting Suppresses Experimental Disease Progression. Cell Death Differ. 2023, 30, 2092–2103. [Google Scholar] [CrossRef]
- Shen, D.; Liu, K.; Wang, H.; Wang, H. Autophagy Modulation in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Clin. Exp. Immunol. 2022, 209, 140. [Google Scholar] [CrossRef]
- Gillen, K.M.; Mubarak, M.; Nguyen, T.D.; Pitt, D. Significance and In Vivo Detection of Iron-Laden Microglia in White Matter Multiple Sclerosis Lesions. Front. Immunol. 2018, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron Accumulation in Senescent Cells Is Coupled with Impaired Ferritinophagy and Inhibition of Ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Adeniyi, P.A.; Gong, X.; MacGregor, E.; Degener-O’Brien, K.; McClendon, E.; Garcia, M.; Romero, O.; Russell, J.; Srivastava, T.; Miller, J.; et al. Ferroptosis of Microglia in Aging Human White Matter Injury. Ann. Neurol. 2023, 94, 1048. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ji, M.; Wu, C.; Zhang, Y.; Ji, S. Targeting Ferroptosis in Neuroimmune and Neurodegenerative Disorders for the Development of Novel Therapeutics. Biomed. Pharmacother. 2024, 176, 116777. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Molina, P.; Almolda, B.; Benseny-Cases, N.; González, B.; Perálvarez-Marín, A.; Castellano, B. Specific Microglial Phagocytic Phenotype and Decrease of Lipid Oxidation in White Matter Areas during Aging: Implications of Different Microenvironments. Neurobiol. Aging 2021, 105, 280–295. [Google Scholar] [CrossRef]
- Quek, H.; Cuní-López, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS Monocyte-Derived Microglia-like Cells Reveal Cytoplasmic TDP-43 Accumulation, DNA Damage, and Cell-Specific Impairment of Phagocytosis Associated with Disease Progression. J. Neuroinflamm. 2022, 19, 58. [Google Scholar] [CrossRef]
- Tam, W.Y.; Ma, C.H.E. Bipolar/Rod-Shaped Microglia Are Proliferating Microglia with Distinct M1/M2 Phenotypes. Sci. Rep. 2014, 4, 7279. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lu, Y.; Do, K.; Mize, T.; Wu, X.; Chen, X.; Chen, J. Validation of Induced Microglia-Like Cells (IMG Cells) for Future Studies of Brain Diseases. Front. Cell Neurosci. 2021, 15, 629279. [Google Scholar] [CrossRef]
- Dai, X.-J.; Li, N.; Yu, L.; Chen, Z.Y.; Hua, R.; Qin, X.; Zhang, Y.M. Activation of BV2 Microglia by Lipopolysaccharide Triggers an Inflammatory Reaction in PC12 Cell Apoptosis through a Toll-like Receptor 4-Dependent Pathway. Cell Stress Chaperones 2014, 20, 321. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, A.J.; Bosco, D.B.; Broussard, E.N.; Ren, Y. In Vitro Phagocytosis of Myelin Debris by Bone Marrow-Derived Macrophages. J. Vis. Exp. 2017, 2017, 56322. [Google Scholar] [CrossRef]
- Bongarzone, E.R.; Pasquini, J.M.; Soto, E.F. Oxidative Damage to Proteins and Lipids of CNS Myelin Produced by in Vitro Generated Reactive Oxygen Species. J. Neurosci. Res. 1995, 41, 213–221. [Google Scholar] [CrossRef]
- Martin, N.P.; Harry, G.J. Imaging Inflammasome Activation in Microglia. Curr. Protoc. 2022, 2, e578. [Google Scholar] [CrossRef] [PubMed]
- Ohgidani, M.; Kato, T.A.; Kanba, S. Introducing Directly Induced Microglia-like (IMG) Cells from Fresh Human Monocytes: A Novel Translational Research Tool for Psychiatric Disorders. Front. Cell Neurosci. 2015, 9, 184. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Dilution | Reference | Secondary Ab | kDa |
|---|---|---|---|---|
| Sod2 | 1:1000 | ab2787145 (Abcam, Cambridge, UK) | Mouse | 20 |
| P-Erk | 1:1000 | ab2809161 (Abcam, Cambridge, UK) | Rabbit | 42–44 |
| Erk ½ total | 1:1000 | ab184699 (Abcam, Cambridge, UK) | Rabbit | 42–44 |
| Catalase | 1:1000 | ab16731 (Abcam, Cambridge, UK) | Rabbit | 60 |
| Cleaved caspase 3 | 1:1000 | 9661(Cell signaling, Danvers, MA, USA) | Rabbit | 32 |
| Gpx4 | 1:1000 | 67763-1-(ProteinTech, Rosemont, IL, USA) | Mouse | 20 |
| Tfr1 | 1:500 | 13-6800 (Thermo Fisher Scientific, Waltham, MA, USA) | Mouse | 84 |
| Acsl4 | 1:1000 | ab155282 (Abcam, Cambridge, UK) | Rabbit | 75 |
| p62 | 1:1000 | 5114 (Cell signaling, Danvers, MA, USA) | Rabbit | 62 |
| Lc3b | 1:1000 | 2775 (Cell signaling, Danvers, MA, USA) | Rabbit | 14–18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santacreu-Vilaseca, M.; Moreno-Magallon, J.; Juanes-Casado, A.; Gil-Sánchez, A.; González-Mingot, C.; Torres, P.; Brieva, L. Novel Cell Models to Study Myelin and Microglia Interactions. Int. J. Mol. Sci. 2025, 26, 2179. https://doi.org/10.3390/ijms26052179
Santacreu-Vilaseca M, Moreno-Magallon J, Juanes-Casado A, Gil-Sánchez A, González-Mingot C, Torres P, Brieva L. Novel Cell Models to Study Myelin and Microglia Interactions. International Journal of Molecular Sciences. 2025; 26(5):2179. https://doi.org/10.3390/ijms26052179
Chicago/Turabian StyleSantacreu-Vilaseca, Marta, Judith Moreno-Magallon, Alba Juanes-Casado, Anna Gil-Sánchez, Cristina González-Mingot, Pascual Torres, and Luis Brieva. 2025. "Novel Cell Models to Study Myelin and Microglia Interactions" International Journal of Molecular Sciences 26, no. 5: 2179. https://doi.org/10.3390/ijms26052179
APA StyleSantacreu-Vilaseca, M., Moreno-Magallon, J., Juanes-Casado, A., Gil-Sánchez, A., González-Mingot, C., Torres, P., & Brieva, L. (2025). Novel Cell Models to Study Myelin and Microglia Interactions. International Journal of Molecular Sciences, 26(5), 2179. https://doi.org/10.3390/ijms26052179