
Design and Synthesis of New 5-Methylisatin Derivatives as Potential CDK2 Inhibitors
 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
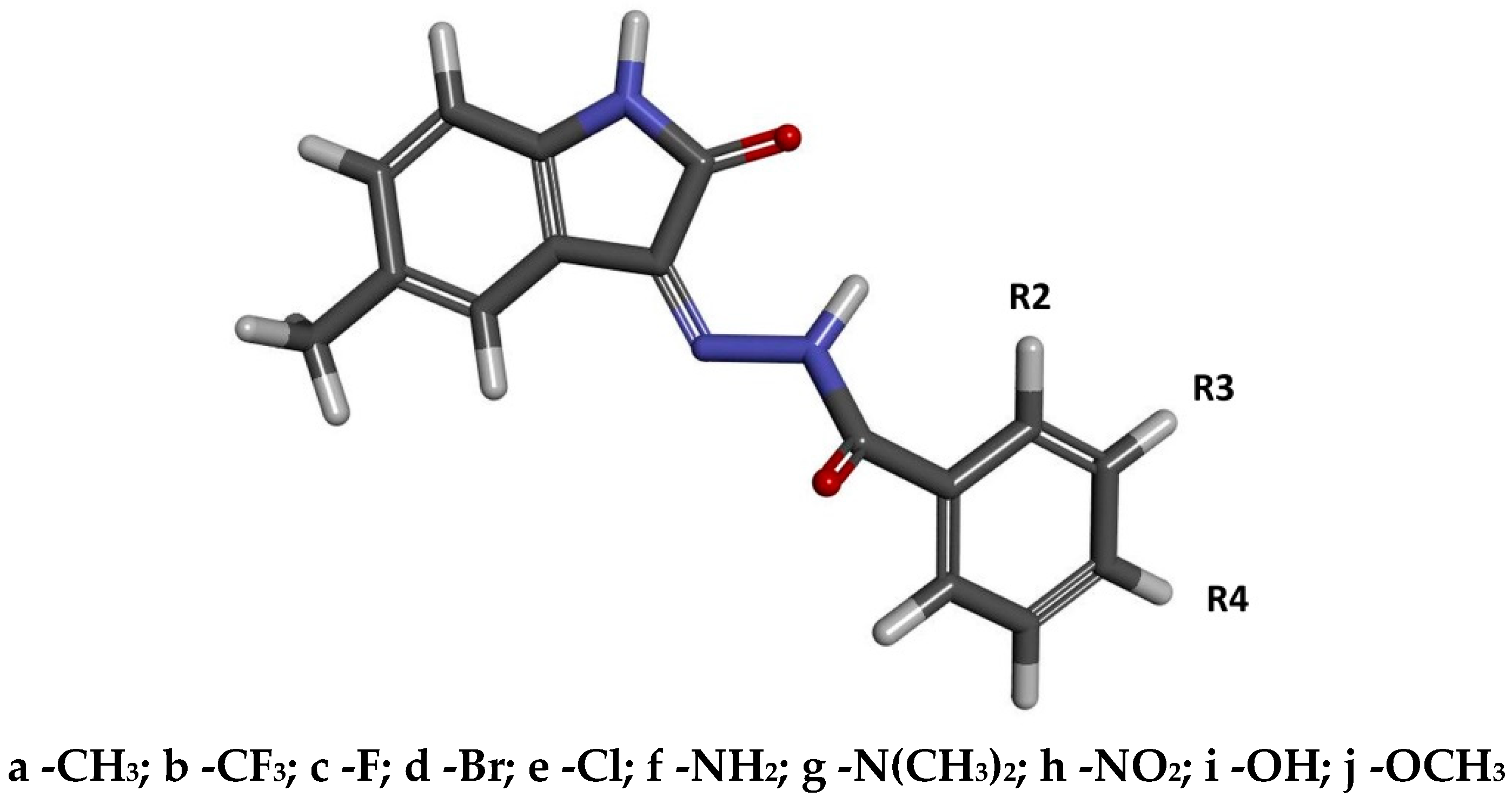
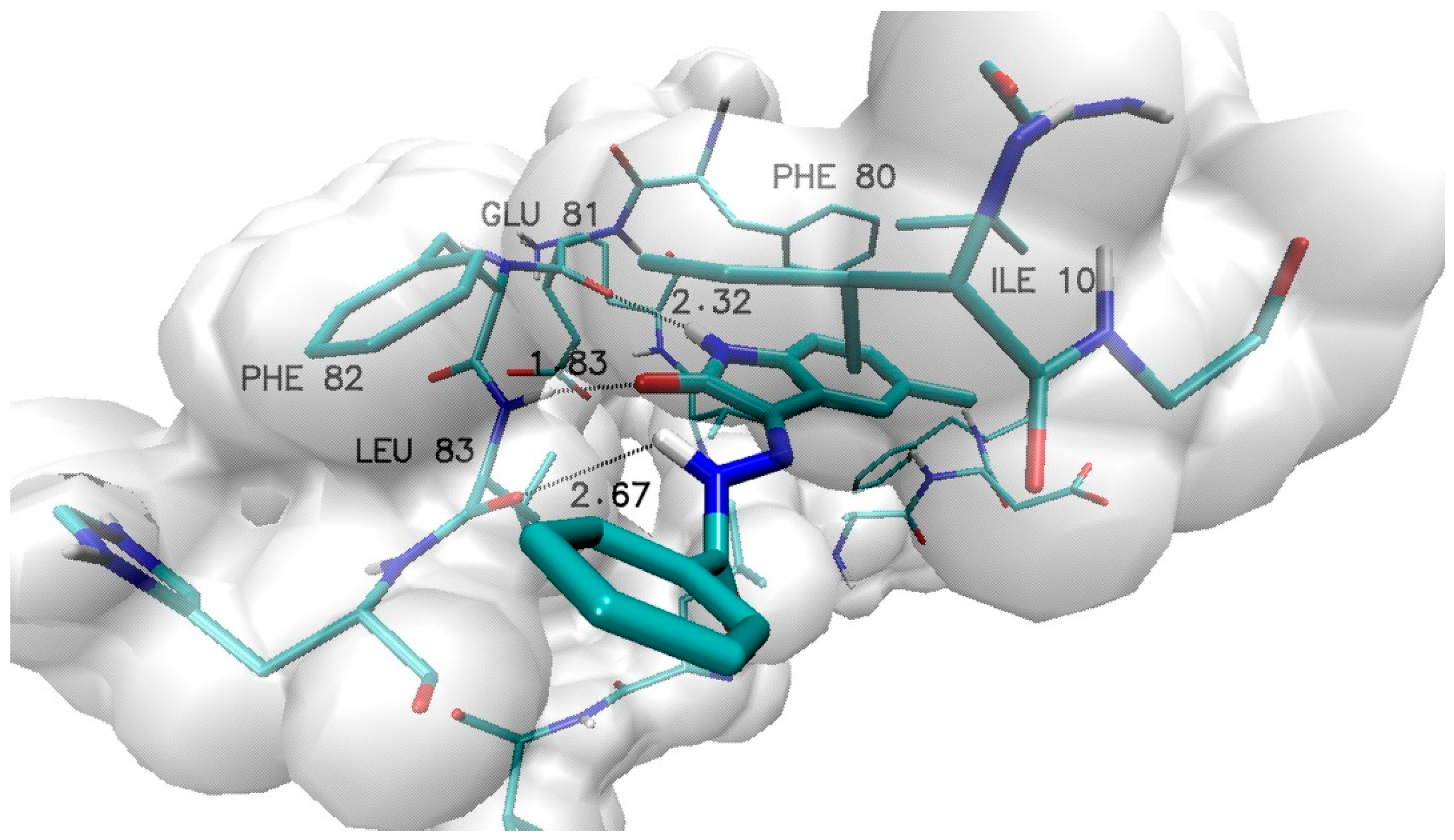
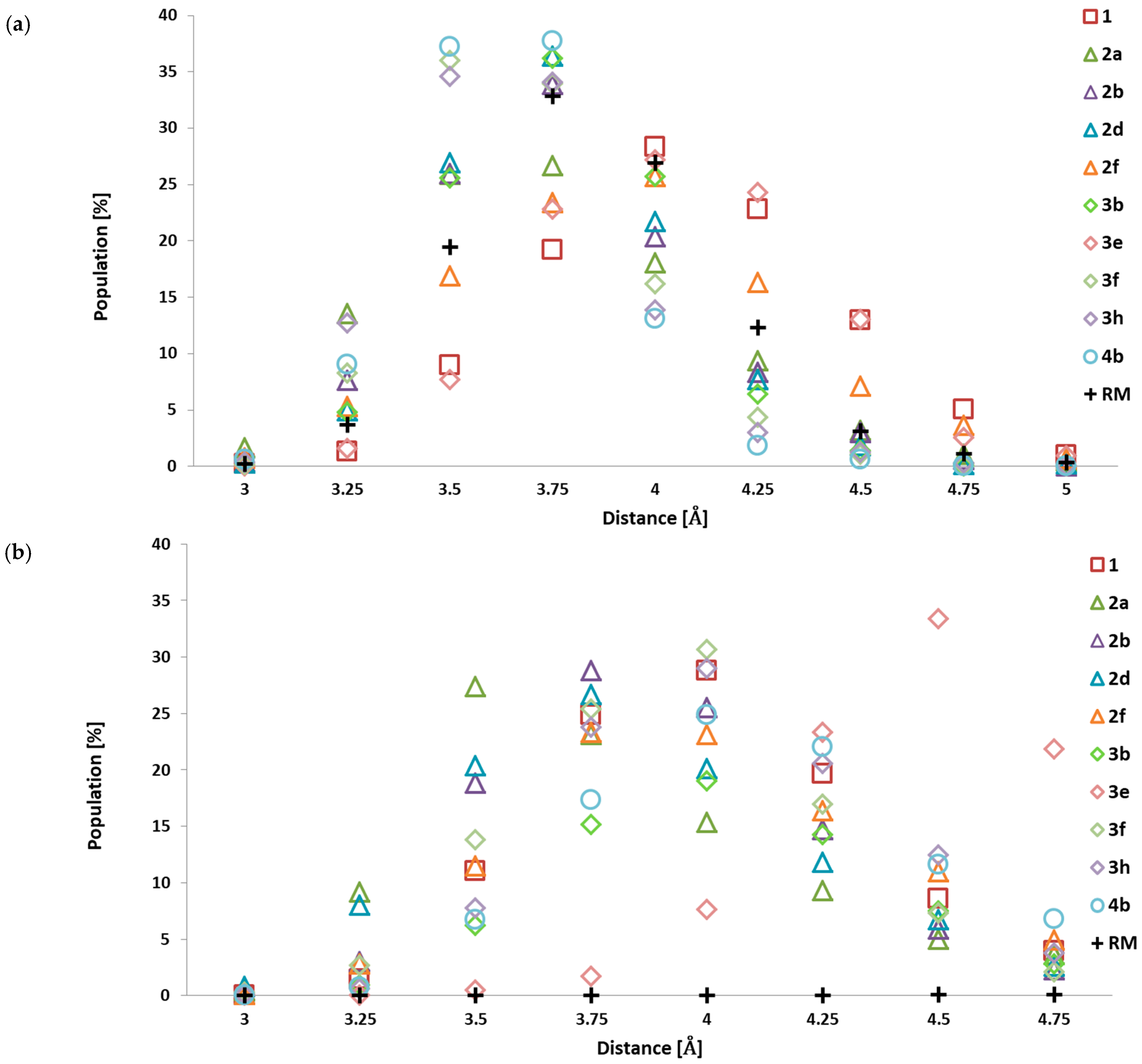
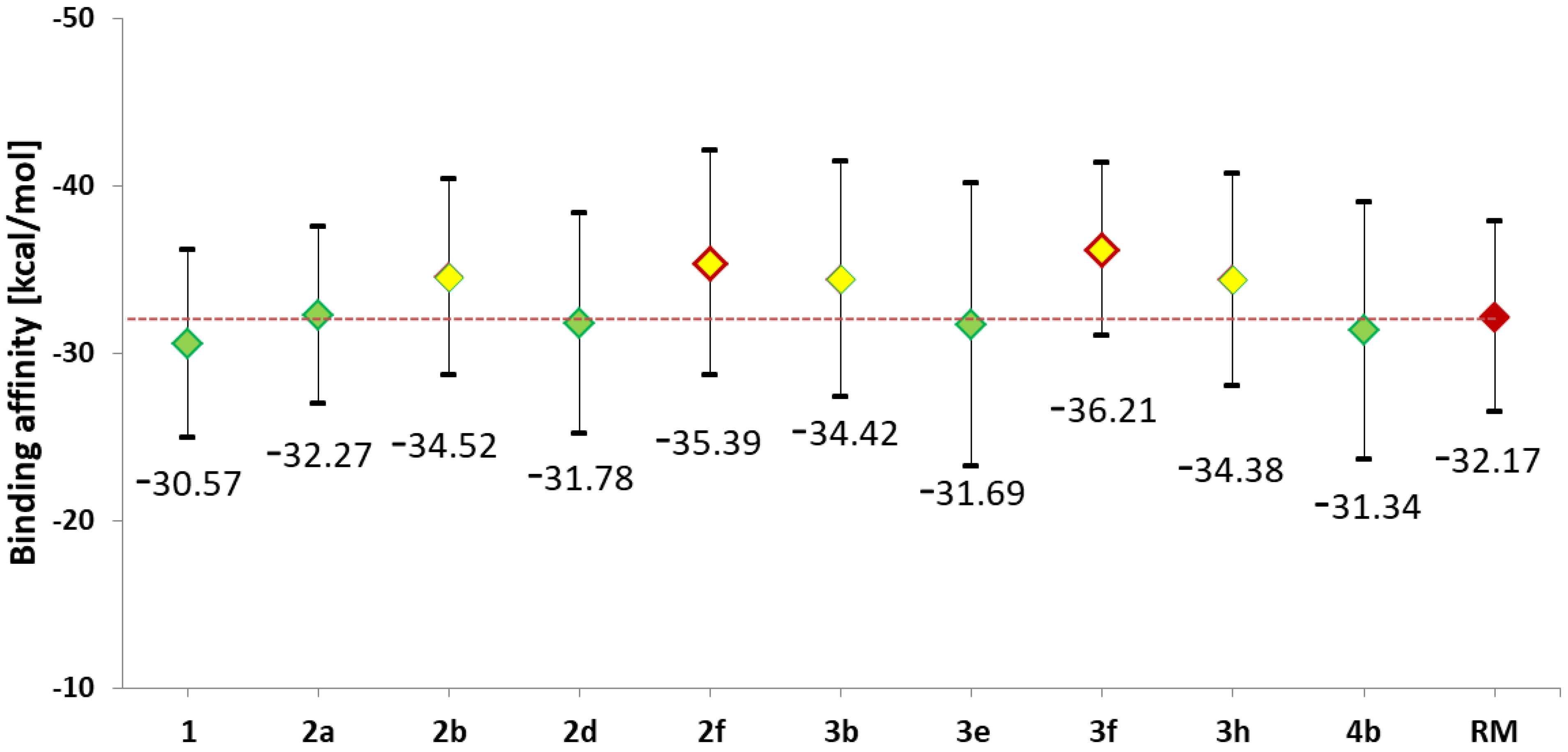
2.1. Design and Computational Analysis of Binding Activity
2.2. The Analysis of ADMET Properties
2.3. Synthesis and NMR Data
2.4. Infrared Spectral Studies
2.5. Spectroscopic Properties
3. Materials and Methods
3.1. Computational Methods
3.1.1. The Docking Procedure and In Silico ADMET Analysis
3.1.2. The Molecular Dynamics Simulations
3.2. Materials
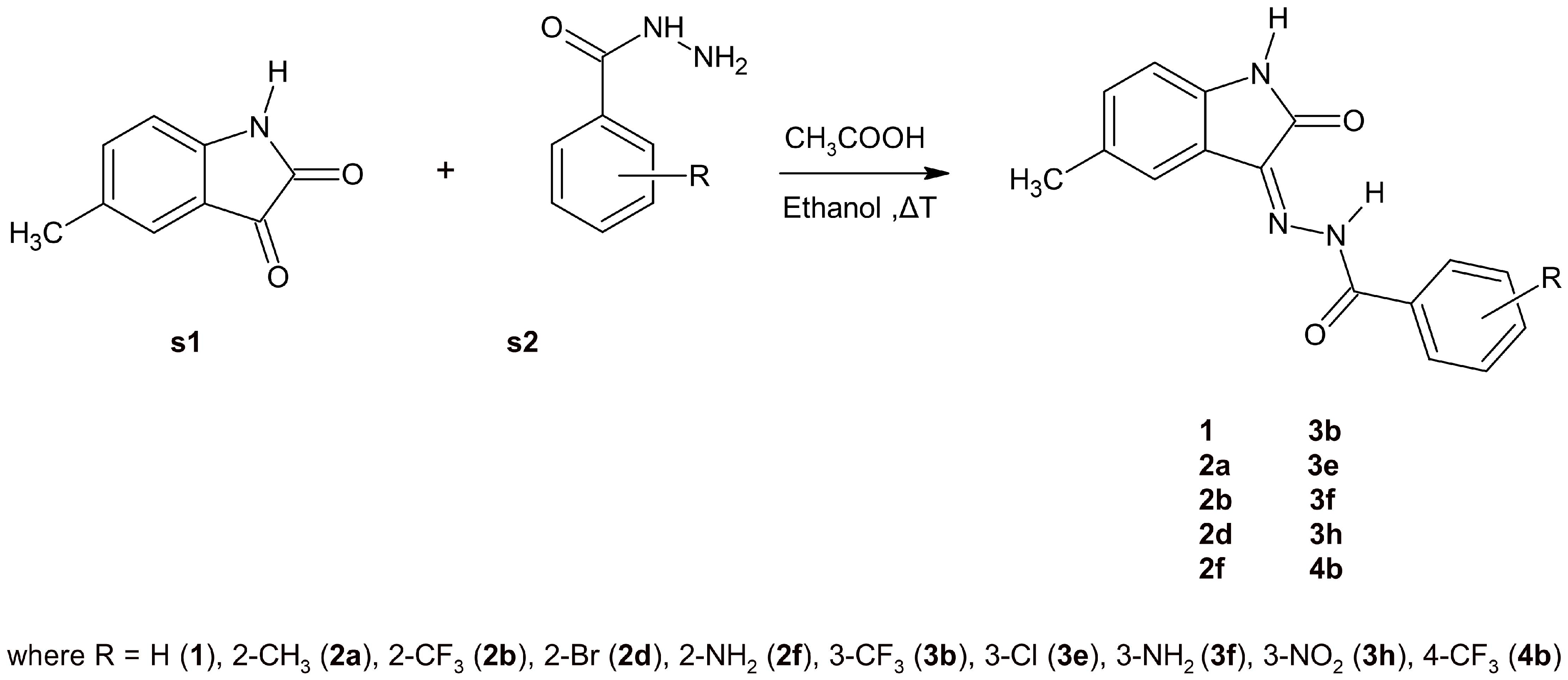
3.3. Synthesis
3.4. Experimental Measurements
3.4.1. NMR Measurements
3.4.2. Elemental Analysis Measurements
3.4.3. UV-VIS Measurements
3.4.4. FTIR Measurements
3.4.5. Calorimetric Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boire, A.; Burke, K.; Cox, T.R.; Guise, T.; Jamal-Hanjani, M.; Janowitz, T.; Kaplan, R.; Lee, R.; Swanton, C.; Vander Heiden, M.G.; et al. Why do patients with cancer die? Nat. Rev. Cancer 2024, 24, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Hjartåker, A.; Weiderpass, E.; Bray, F. Cancer Mortality. In International Encyclopedia of Public Health; Elsevier: Amsterdam, The Netherlands, 2017; pp. 369–380. [Google Scholar]
- Morgan, D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Dong, P.; Gassler, N.; Taheri, M.; Baniahmad, A.; Dilmaghani, N.A. A review on the role of cyclin dependent kinases in cancers. Cancer Cell Int. 2022, 22, 325. [Google Scholar] [CrossRef] [PubMed]
- Ferraz de Paiva, R.E.; Vieira, E.G.; Rodrigues da Silva, D.; Wegermann, C.A.; Costa Ferreira, A.M. Anticancer Compounds Based on Isatin-Derivatives: Strategies to Ameliorate Selectivity and Efficiency. Front. Mol. Biosci. 2021, 7, 511. [Google Scholar] [CrossRef] [PubMed]
- Lashen, A.; Alqahtani, S.; Shoqafi, A.; Algethami, M.; Jeyapalan, J.N.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. Clinicopathological Significance of Cyclin-Dependent Kinase 2 (CDK2) in Ductal Carcinoma In Situ and Early-Stage Invasive Breast Cancers. Int. J. Mol. Sci. 2024, 25, 5053. [Google Scholar] [CrossRef]
- Sabnis, R.W. Novel CDK2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2020, 11, 2346–2347. [Google Scholar] [CrossRef] [PubMed]
- Chenette, E.J. A key role for CDK2. Nat. Rev. Cancer 2010, 10, 84. [Google Scholar] [CrossRef]
- Sabt, A.; Eldehna, W.M.; Al-Warhi, T.; Alotaibi, O.J.; Elaasser, M.M.; Suliman, H.; Abdel-Aziz, H.A. Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: Synthesis, biological evaluation and in silico insights. J. Enzym. Inhib. Med. Chem. 2020, 35, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Bentley, J.; Arris, C.E.; Boyle, F.T.; Curtin, N.J.; Endicott, J.A.; Gibson, A.E.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat. Struct. Biol. 2002, 9, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Coxon, C.R.; Anscombe, E.; Harnor, S.J.; Martin, M.P.; Carbain, B.; Golding, B.T.; Hardcastle, I.R.; Harlow, L.K.; Korolchuk, S.; Matheson, C.J.; et al. Cyclin-Dependent Kinase (CDK) Inhibitors: Structure–Activity Relationships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopurines. J. Med. Chem. 2017, 60, 1746–1767. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S. Inhibition of Cyclin-Dependent Kinases by Purine Analogues. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef]
- Jorda, R.; Havlíček, L.; McNae, I.W.; Walkinshaw, M.D.; Voller, J.; Šturc, A.; Navrátilová, J.; Kuzma, M.; Mistrík, M.; Bártek, J.; et al. Pyrazolo[4,3-d]pyrimidine Bioisostere of Roscovitine: Evaluation of a Novel Selective Inhibitor of Cyclin-Dependent Kinases with Antiproliferative Activity. J. Med. Chem. 2011, 54, 2980–2993. [Google Scholar] [CrossRef]
- Liu, J.-J.; Daniewski, I.; Ding, Q.; Higgins, B.; Ju, G.; Kolinsky, K.; Konzelmann, F.; Lukacs, C.; Pizzolato, G.; Rossman, P.; et al. Pyrazolobenzodiazepines: Part I. Synthesis and SAR of a potent class of kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5984–5987. [Google Scholar] [CrossRef] [PubMed]
- Brasca, M.G.; Amboldi, N.; Ballinari, D.; Cameron, A.; Casale, E.; Cervi, G.; Colombo, M.; Colotta, F.; Croci, V.; D’Alessio, R.; et al. Identification of N,1,4,4-Tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro-1 H-pyrazolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a Potent, Orally Available Cyclin Dependent Kinase Inhibitor. J. Med. Chem. 2009, 52, 5152–5163. [Google Scholar] [CrossRef]
- Finlay, M.R.V.; Acton, D.G.; Andrews, D.M.; Barker, A.J.; Dennis, M.; Fisher, E.; Graham, M.A.; Green, C.P.; Heaton, D.W.; Karoutchi, G.; et al. Imidazole piperazines: SAR and development of a potent class of cyclin-dependent kinase inhibitors with a novel binding mode. Bioorg. Med. Chem. Lett. 2008, 18, 4442–4446. [Google Scholar] [CrossRef]
- Lin, S.-F.; Lin, J.-D.; Hsueh, C.; Chou, T.-C.; Wong, R.J. Activity of roniciclib in medullary thyroid cancer. Oncotarget 2018, 9, 28030–28041. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ramarao, T.A.; Samanta, P.K.; Jha, A.; Satpati, P.; Sen, A. Triazole based isatin derivatives as potential inhibitor of key cancer promoting kinases- insight from electronic structure, docking and molecular dynamics simulations. J. Mol. Graph. Model. 2021, 107, 107944. [Google Scholar] [CrossRef]
- Shin, E.-K.; Kim, J.-K. Indirubin derivative E804 inhibits angiogenesis. BMC Cancer 2012, 12, 164. [Google Scholar] [CrossRef]
- Bramson, H.N.; Holmes, W.D.; Hunter, R.N.; Lackey, K.E.; Lovejoy, B.; Luzzio, M.J.; Montana, V.; Rocque, W.J.; Rusnak, D.; Shewchuk, L.; et al. Oxindole-based inhibitors of cyclin-dependent kinase 2 (CDK2): Design, synthesis, enzymatic activities, and X-ray crystallographic analysis. J. Med. Chem. 2001, 44, 4339–4358. [Google Scholar] [CrossRef] [PubMed]
- Czeleń, P. Inhibition mechanism of CDK-2 and GSK-3β by a sulfamoylphenyl derivative of indoline—A molecular dynamics study. J. Mol. Model. 2017, 23, 230. [Google Scholar] [CrossRef]
- Czeleń, P.; Szefler, B. Molecular dynamics study of the inhibitory effects of ChEMBL474807 on the enzymes GSK-3β and CDK-2. J. Mol. Model. 2015, 21, 74. [Google Scholar] [CrossRef] [PubMed]
- Czeleń, P. Molecular dynamics study on inhibition mechanism of CDK-2 and GSK-3β by CHEMBL272026 molecule. Struct. Chem. 2016, 27, 1807–1818. [Google Scholar] [CrossRef]
- Bharathi Dileepan, A.G.; Daniel Prakash, T.; Ganesh Kumar, A.; Shameela Rajam, P.; Violet Dhayabaran, V.; Rajaram, R. Isatin based macrocyclic Schiff base ligands as novel candidates for antimicrobial and antioxidant drug design: In vitro DNA binding and biological studies. J. Photochem. Photobiol. B Biol. 2018, 183, 191–200. [Google Scholar] [CrossRef]
- Guo, H. Isatin derivatives and their anti-bacterial activities. Eur. J. Med. Chem. 2019, 164, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Patil, V.M.; Firke, S.D.; Ambhore, J.P.; Ansari, I.A.; Patel, H.M.; Shinde, S.D.; Pasupuleti, V.R.; Hassan, M.I.; Adnan, M.; et al. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals 2022, 15, 272. [Google Scholar] [CrossRef]
- Nath, R.; Pathania, S.; Grover, G.; Akhtar, M.J. Isatin containing heterocycles for different biological activities: Analysis of structure activity relationship. J. Mol. Struct. 2020, 1222, 128900. [Google Scholar] [CrossRef]
- Elsaman, T.; Mohamed, M.S.; Eltayib, E.M.; Abdel-aziz, H.A.; Abdalla, A.E.; Munir, M.U.; Mohamed, M.A. Isatin derivatives as broad-spectrum antiviral agents: The current landscape. Med. Chem. Res. 2022, 31, 244–273. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abou-Seri, S.M.; Supuran, C.T. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur. J. Med. Chem. 2018, 157, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Cheke, R.S.; Firke, S.D.; Patil, R.R.; Bari, S.B. ISATIN: New Hope Against Convulsion. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 76–101. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef]
- Czeleń, P.; Skotnicka, A.; Szefler, B. Designing and Synthesis of New Isatin Derivatives as Potential CDK2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 8046. [Google Scholar] [CrossRef] [PubMed]
- Czeleń, P.; Szefler, B. The Oxindole Derivatives, New Promising GSK-3β Inhibitors as One of the Potential Treatments for Alzheimer’s Disease—A Molecular Dynamics Approach. Biology 2021, 10, 332. [Google Scholar] [CrossRef]
- Czeleń, P. Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy. Symmetry 2019, 11, 974. [Google Scholar] [CrossRef]
- Czeleń, P.; Jeliński, T.; Skotnicka, A.; Szefler, B.; Szupryczyński, K. ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes. Biomedicines 2023, 11, 3019. [Google Scholar] [CrossRef]
- Al-Salem, H.S.; Arifuzzaman, M.; Alkahtani, H.M.; Abdalla, A.N.; Issa, I.S.; Alqathama, A.; Albalawi, F.S.; Rahman, A.F.M.M. A Series of Isatin-Hydrazones with Cytotoxic Activity and CDK2 Kinase Inhibitory Activity: A Potential Type II ATP Competitive Inhibitor. Molecules 2020, 25, 4400. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Li, Y.; Song, Y.; Li, D.; Sun, H.; Hou, T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminform. 2016, 8, 6. [Google Scholar] [CrossRef]
- Wang, S.; Sun, H.; Liu, H.; Li, D.; Li, Y.; Hou, T. ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol. Pharm. 2016, 13, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R.J. Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-N.; Dong, J.; Deng, Y.-H.; Zhu, M.-F.; Wen, M.; Yao, Z.-J.; Lu, A.-P.; Wang, J.-B.; Cao, D.-S. ADME Properties Evaluation in Drug Discovery: Prediction of Caco-2 Cell Permeability Using a Combination of NSGA-II and Boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, A.; Hegde, M.; Kumar, S.; Gopalakrishnan, V.; Bhatelia, K.D.; Ananthaswamy, K.; Ramareddy, S.A.; De Clercq, E.; Choudhary, B.; Schols, D.; et al. Synthesis and evaluation of the biological activity of N′-[2-oxo-1,2 dihydro-3H-indol-3-ylidene] benzohydrazides as potential anticancer agents. RSC Adv. 2015, 5, 45492–45501. [Google Scholar] [CrossRef]
- Debnath, K.; Pathak, S.; Pramanik, A. Facile synthesis of ninhydrin and isatin based hydrazones in water using PEG-OSO3H as a highly efficient and homogeneous polymeric acid-surfactant combined catalyst. Tetrahedron Lett. 2013, 54, 4110–4115. [Google Scholar] [CrossRef]
- Haj Mohammad Ebrahim Tehrani, K.; Hashemi, M.; Hassan, M.; Kobarfard, F.; Mohebbi, S. Synthesis and antibacterial activity of Schiff bases of 5-substituted isatins. Chin. Chem. Lett. 2016, 27, 221–225. [Google Scholar] [CrossRef]
- Emami, S.; Valipour, M.; Kazemi Komishani, F.; Sadati-Ashrafi, F.; Rasoulian, M.; Ghasemian, M.; Tajbakhsh, M.; Honarchian Masihi, P.; Shakiba, A.; Irannejad, H.; et al. Synthesis, in silico, in vitro and in vivo evaluations of isatin aroylhydrazones as highly potent anticonvulsant agents. Bioorg. Chem. 2021, 112, 104943. [Google Scholar] [CrossRef] [PubMed]
- Hunoor, R.S.; Patil, B.R.; Badiger, D.S.; Chandrashekhar, V.M.; Muchchandi, I.S.; Gudasi, K.B. Co(II), Ni(II), Cu(II) and Zn(II) complexes of isatinyl-2-aminobenzoylhydrazone: Synthesis, characterization and anticancer activity. Appl. Organomet. Chem. 2015, 29, 101–108. [Google Scholar] [CrossRef]
- Davies, T.G.; Tunnah, P.; Meijer, L.; Marko, D.; Eisenbrand, G.; Endicott, J.A.; Noble, M.E. Inhibitor binding to active and inactive CDK2: The crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure 2001, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.V.; Potemkin, V.A.; Grishina, M.A.; Belik, A.V. A Method for Multiconformational Modeling of the Three-Dimensional Shape of a Molecule. J. Struct. Chem. 2002, 43, 1033–1039. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Adelman, S.A. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14 2014, University of California, San Francisco. Available online: https://orbilu.uni.lu/handle/10993/16614 (accessed on 23 February 2025).
- Kelava, A.; Moosbrugger, H.; Dimitruk, P.; Schermelleh-Engel, K. Methodology: European Journal of Research Methods for the Behavioral and Social Sciences. Methodology 2008, 4, 51–66. [Google Scholar] [CrossRef]
- Stoline, M.R. The Status of Multiple Comparisons: Simultaneous Estimation of All Pairwise Comparisons in One-Way ANOVA Designs. Am. Stat. 1981, 35, 134. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Group | Name | Binding Affinity [kcal/mol] | IC [nM] | Name | Binding Affinity [kcal/mol] | IC [nM] | Name | Binding Affinity [kcal/mol] | IC [nM] |
|---|---|---|---|---|---|---|---|---|---|
| -H | 1 | −9.18 (0.04) | 186.63 | --- | --- | --- | --- | --- | --- |
| -CH3 | 2a | −9.60 (0.00) | 91.86 | 3a | −9.50 (0.00) | 108.74 | 4a | −9.44 (0.05) | 120.33 |
| -CF3 | 2b | −9.80 (0.00) | 65.54 | 3b | −9.84 (0.05) | 61.26 | 4b | −9.70 (0.00) | 77.59 |
| -F | 2c | −9.40 (0.00) | 128.74 | 3c | −9.50 (0.00) | 108.74 | 4c | −9.30 (0.00) | 152.41 |
| -Br | 2d | −9.58 (0.04) | 95.01 | 3d | −9.50 (0.00) | 108.74 | 4d | −9.40 (0.00) | 128.74 |
| -Cl | 2e | −9.50 (0.00) | 108.74 | 3e | −9.56 (0.05) | 98.27 | 4e | −9.32 (0.04) | 147.35 |
| -NH2 | 2f | −9.60 (0.00) | 91.86 | 3f | −9.64 (0.05) | 85.86 | 4f | −9.10 (0.00) | 213.61 |
| -N(CH3)2 | 2g | −8.30 (0.00) | 824.17 | 3g | −9.10 (0.00) | 213.61 | 4g | −9.20 (0.00) | 180.43 |
| -NO2 | 2h | −9.00 (0.00) | 252.88 | 3h | −9.50 (0.00) | 108.74 | 4h | −9.24 (0.05) | 168.65 |
| -OH | 2i | −9.20 (0.00) | 180.43 | 3i | −9.20 (0.00) | 180.43 | 4i | −9.10 (0.00) | 213.61 |
| -OCH3 | 2j | −8.98 (0.00) | 261.56 | 3j | −9.20 (0.00) | 180.43 | 4j | −9.20 (0.00) | 180.43 |
| Name | Distances [Å] | ||||
|---|---|---|---|---|---|
| GLU 81 | LEU 83 H | LEU 83 O | Phe80 | Phe82 | |
| 1 | 2.32 | 1.83 | 2.67 | 4.08 | 4.06 |
| 2a | 2.15 | 1.88 | 3.23 | 3.57 | 4.07 |
| 2b | 2.15 | 1.90 | 3.42 | 3.82 | 4.01 |
| 2d | 2.13 | 1.85 | 3.32 | 3.87 | 4.06 |
| 2f | 2.21 | 1.88 | 2.96 | 3.58 | 4.11 |
| 3b | 2.14 | 1.87 | 3.28 | 3.57 | 4.36 |
| 3e | 2.19 | 1.86 | 3.05 | 3.90 | 4.08 |
| 3f | 2.24 | 1.82 | 2.80 | 4.04 | 4.03 |
| 3h | 2.43 | 1.84 | 2.53 | 4.23 | 3.69 |
| 4b | 2.21 | 1.84 | 2.81 | 3.95 | 3.88 |
| RM | 2.41 | 2.04 | ---- | 3.70 | 5.17 |
| Name | Ligand | CDK2 | ||||||
|---|---|---|---|---|---|---|---|---|
| 80 ns | Last 60 ns | 80 ns | Last 60 ns | |||||
| RMSD | SD | RMSD | SD | RMSD | SD | RMSD | SD | |
| 1 | 0.33 | 0.10 | 0.33 | 0.10 | 2.44 | 0.28 | 2.56 | 0.12 |
| 2a | 1.27 | 0.41 | 1.45 | 0.06 | 2.57 | 0.36 | 2.70 | 0.26 |
| 2b | 0.89 | 0.17 | 0.91 | 0.16 | 2.49 | 0.40 | 2.68 | 0.19 |
| 2d | 0.45 | 0.16 | 0.45 | 0.17 | 2.27 | 0.19 | 2.29 | 0.15 |
| 2f | 0.77 | 0.16 | 0.79 | 0.17 | 2.67 | 0.33 | 2.77 | 0.28 |
| 3b | 1.53 | 0.59 | 1.82 | 0.35 | 2.31 | 0.31 | 2.43 | 0.19 |
| 3e | 0.63 | 0.22 | 0.65 | 0.22 | 2.52 | 0.34 | 2.63 | 0.26 |
| 3f | 0.25 | 0.07 | 0.24 | 0.06 | 2.16 | 0.17 | 2.19 | 0.11 |
| 3h | 0.66 | 0.15 | 0.67 | 0.15 | 2.35 | 0.30 | 2.47 | 0.19 |
| 4a | 0.91 | 0.29 | 0.96 | 0.30 | 2.41 | 0.37 | 2.50 | 0.34 |
| RM | 1.16 | 0.25 | 1.24 | 0.12 | 2.49 | 0.24 | 2.58 | 0.15 |
| Interactions | Population % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Σ | 1.6 Å | 1.8 Å | 2 Å | 2.2 Å | 2.4 Å | 2.6 Å | 2.8 Å | 3 Å | |
| 1 | |||||||||
| Ligand (H1) … (O) GLU 81 | 100 | 1.94 | 48.31 | 41.44 | 7.56 | 0.69 | 0.06 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100 | 0.88 | 26.75 | 46.13 | 19.50 | 5.38 | 1.25 | 0.13 | 0.00 |
| Ligand (H5) … (O) LEU 83 | 20.6 | 0.00 | 0.19 | 0.31 | 0.75 | 1.25 | 3.44 | 4.94 | 9.69 |
| 2a | |||||||||
| Ligand (H1) … (O) GLU 81 | 100 | 1.94 | 48.94 | 40.94 | 7.63 | 0.50 | 0.06 | 0.00 | 0.00 |
| Ligand (O2) … (HN) LEU 83 | 99.56 | 0.25 | 13.69 | 30.94 | 28.56 | 16.31 | 5.56 | 3.00 | 1.25 |
| 2b | |||||||||
| Ligand (H1) … (O) GLU 81 | 100 | 1.19 | 41.31 | 44.00 | 11.37 | 1.81 | 0.31 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100 | 1 | 29 | 46.8125 | 18.125 | 4 | 0.9375 | 0.0625 | 0.0625 |
| Ligand (H5) … (O) LEU 83 | 55.5 | 0.44 | 10.62 | 18.56 | 11.00 | 4.31 | 2.62 | 3.75 | 4.19 |
| 2d | |||||||||
| Ligand (H1) … (O) GLU 81 | 100 | 2.25 | 49.44 | 41.31 | 6.37 | 0.62 | 0.00 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100 | 0.12 | 16.19 | 39.94 | 27.94 | 10.81 | 3.87 | 1.00 | 0.12 |
| 2f | |||||||||
| Ligand (H1) … (O) GLU 81 | 100 | 2.69 | 48.00 | 40.19 | 8.06 | 0.81 | 0.25 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 99.94 | 0.75 | 29.81 | 45.25 | 16.75 | 5.56 | 1.44 | 0.19 | 0.19 |
| Ligand (H) … (O) ILE10 | 62.44 | 0.13 | 7.63 | 15.25 | 12.38 | 10.19 | 6.38 | 5.00 | 5.50 |
| Ligand (NH) … (O2) Ligand | 98.13 | 0.00 | 7.44 | 35.81 | 30.94 | 14.63 | 5.69 | 2.31 | 1.31 |
| 3b | |||||||||
| Ligand (H1) … (O) GLU 81 | 100.00 | 1.25 | 41.31 | 44.75 | 10.88 | 1.63 | 0.13 | 0.06 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100.00 | 1.06 | 30.69 | 42.13 | 18.69 | 5.44 | 1.56 | 0.44 | 0.00 |
| Ligand (H5) … (O) LEU 83 | 46.56 | 0.06 | 4.44 | 10.69 | 7.38 | 6.19 | 4.75 | 5.63 | 7.44 |
| 3e | |||||||||
| Ligand (H3) … (O) GLU 81 | 100.00 | 2.00 | 49.00 | 41.63 | 6.50 | 0.88 | 0.00 | 0.00 | 0.00 |
| Ligand (O2) … (HN) LEU 83 | 100.00 | 0.88 | 34.19 | 46.06 | 15.19 | 3.06 | 0.63 | 0.00 | 0.00 |
| Ligand (H5) … (O) LEU 83 | 39.44 | 0.00 | 0.69 | 2.06 | 2.88 | 3.50 | 6.44 | 9.56 | 14.31 |
| 3f | |||||||||
| Ligand (H1) … (O) GLU 81 | 100.00 | 1.63 | 43.88 | 44.19 | 9.56 | 0.63 | 0.13 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100.00 | 0.25 | 14.69 | 44.00 | 27.69 | 10.25 | 2.56 | 0.44 | 0.13 |
| Ligand (NH) … (O) HIE 84 | 97.06 | 0.44 | 14.88 | 33.94 | 22.13 | 11.81 | 7.19 | 4.31 | 2.38 |
| Ligand (NH) … (O) LEU 83 | 99.56 | 0.50 | 18.13 | 41.50 | 25.56 | 8.94 | 3.56 | 1.19 | 0.19 |
| Ligand (H5) … (O1) Ligand | 100.00 | 0.00 | 2.25 | 23.13 | 41.38 | 23.75 | 7.88 | 1.38 | 0.00 |
| 3h | |||||||||
| Ligand (H1) … (O) GLU 81 | 100.00 | 1.00 | 42.81 | 43.88 | 10.31 | 1.56 | 0.44 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100.00 | 1.00 | 32.25 | 45.94 | 15.75 | 3.81 | 0.94 | 0.13 | 0.19 |
| Ligand (H5) … (O) LEU 83 | 44.13 | 0.00 | 0.81 | 4.50 | 4.31 | 7.00 | 7.56 | 9.38 | 10.56 |
| 4b | |||||||||
| Ligand (H1) … (O) GLU 81 | 100.00 | 2.06 | 45.63 | 42.69 | 8.31 | 1.13 | 0.19 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 100.00 | 1.38 | 32.69 | 42.38 | 18.06 | 4.00 | 1.25 | 0.19 | 0.06 |
| Ligand (H5) … (O) LEU 83 | 30.38 | 0.00 | 0.81 | 2.44 | 3.63 | 4.31 | 4.31 | 5.69 | 9.19 |
| RM | |||||||||
| Ligand (H1) … (O) GLU 81 | 99.94 | 1.31 | 39.44 | 44.25 | 12.94 | 1.75 | 0.25 | 0.00 | 0.00 |
| Ligand (O1) … (HN) LEU 83 | 98.94 | 0.13 | 5.69 | 23.81 | 27.25 | 19.75 | 12.06 | 7.13 | 3.13 |
| Name | MW a | nHD b | nHA c | TPSA d | LogP e | LogD f | LogS g | BBB h | Caco-2 i | MDCK j | HIA k | hERG l |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 279.10 | 2 | 5 | 70.56 | 3.088 | 2.737 | −4.669 | 0.337 | −4.893 | 1.25 × 10−5 | 0.011 | 0.051 |
| 2a | 293.12 | 2 | 5 | 70.56 | 3.450 | 2.840 | −5.142 | 0.270 | −4.855 | 1.11 × 10−5 | 0.005 | 0.038 |
| 2b | 347.09 | 2 | 5 | 70.56 | 3.594 | 2.991 | −5.05 | 0.183 | −4.880 | 1.84 × 10−5 | 0.004 | 0.026 |
| 2d | 357.01 | 2 | 5 | 70.56 | 3.613 | 2.871 | −5.339 | 0.452 | −4.804 | 1.51 × 10−5 | 0.006 | 0.064 |
| 2f | 294.11 | 4 | 6 | 96.58 | 3.118 | 2.591 | −5.167 | 0.867 | −5.287 | 8.09 × 10−6 | 0.008 | 0.056 |
| 3b | 347.09 | 2 | 5 | 70.56 | 3.848 | 3.105 | −5.402 | 0.192 | −4.919 | 1.65 × 10−5 | 0.005 | 0.058 |
| 3e | 313.06 | 2 | 5 | 70.56 | 3.777 | 2.839 | −5.205 | 0.244 | −4.841 | 1.49 × 10−5 | 0.008 | 0.089 |
| 3f | 294.11 | 4 | 6 | 96.58 | 2.711 | 2.153 | −4.584 | 0.761 | −5.486 | 6.13 × 10−6 | 0.009 | 0.060 |
| 3h | 324.09 | 2 | 8 | 113.7 | 3.111 | 2.409 | −4.900 | 0.063 | −4.991 | 2.57 × 10−5 | 0.012 | 0.300 |
| 4a | 347.09 | 2 | 5 | 70.56 | 3.881 | 3.111 | −5.512 | 0.140 | −4.880 | 1.51 × 10−5 | 0.004 | 0.062 |
| RM | 317.01 | 1 | 4 | 53.82 | 4.462 | 2.795 | −6.693 | 0.793 | −4.793 | 2.09 × 10−5 | 0.003 | 0.001 |
| No. | Substituent | cis Isomer (%) | |
|---|---|---|---|
| 1 | H | 100 | - |
| 2a | 2-CH3 | 100 | - |
| 2b | 2-CF3 | 22 | 0.28 |
| 2d | 2-Br | 28 | 0.39 |
| 2f | 2-NH2 | 49 | 0.96 |
| 3b | 3-CF3 | 61 | 1.56 |
| 3e | 3-Cl | 49 | 0.96 |
| 3f | 3-NH2 | 69 | 2.23 |
| 3h | 3-NO2 | 43 | 0.75 |
| 4b | 4-CF3 | 44 | 0.78 |
| Compound | ν(–NH2) | ν(N–H) | ν(C=O) | ν(C=N) | ν(N=O) | ||
|---|---|---|---|---|---|---|---|
| νasym | νsym | Lactam | Hydrazide | ||||
| 1 | - | - | 3247 | 1697 | 1674 | 1525 | 1480 |
| 2a | - | - | 3254 | 1683 | 1628 | 1524 | 1482 |
| 2b | - | - | 3265 | 1727 | 1684 | 1512 | 1314 |
| 2d | - | - | 3200 | 1740 | 1619 | 1500 | 1291 |
| 2f | 3483 | 3347 | 3177 | 1716 | 1627 | 1487 | 1318 |
| 3b | - | - | 3282 | 1714 | 1674 | 1498 | 1330 |
| 3e | - | - | 3278 | 1731 | 1677 | 1484 | 1321 |
| 3f | 3449 | 3367 | 3298 | 1680 | 1622 | 1543 | 1314 |
| 3h | - | - | 3285 | 1731 | 1678 | 1532 | 1347 |
| 4b | - | - | 3247 | 1728 | 1662 | 1483 | 1315 |
| No. | Substituent | Solvent | λab (nm) | ε (×104, M−1·cm−1) |
|---|---|---|---|---|
| 1 | H | DCM | 345 | 1.51 |
| MeOH | 328 | 2.16 | ||
| MeCN | 327 | 2.02 | ||
| 2a | 2-CH3 | DCM | 340 | 1.38 |
| MeOH | 326 | 1.84 | ||
| MeCN | 324 | 1.94 | ||
| 2b | 2-CF3 | DCM | 316 | 1.76 |
| MeOH | 320 | 1.63 | ||
| MeCN | 316 | 1.65 | ||
| 2d | 2-Br | DCM | 320 | 1.11 |
| MeOH | 324 | 1.36 | ||
| MeCN | 319 | 1.73 | ||
| 2f | 2-NH2 | DCM | 324, 383 | 1.10, 1.10 |
| MeOH | 325, 386 | 1.67, 1.66 | ||
| MeCN | 322, 387 | 1.95, 1.92 | ||
| 3b | 3-CF3 | DCM | 327 | 1.86 |
| MeOH | 326 | 1.42 | ||
| MeCN | 328 | 1.83 | ||
| 3e | 3-Cl | DCM | 327 | 1.70 |
| MeOH | 329 | 1.63 | ||
| MeCN | 325 | 1.70 | ||
| 3f | 3-NH2 | DCM | 346 | 1.32 |
| MeOH | 328 | 2.09 | ||
| MeCN | 325 | 1.08 | ||
| 3h | 3-NO2 | DCM | 325 | 1.53 |
| MeOH | 329 | 1.46 | ||
| MeCN | 324 | 1.18 | ||
| 4b | 4-CF3 | DCM | 325 | 1.64 |
| MeOH | 328 | 1.32 | ||
| MeCN | 325 | 1.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czeleń, P.; Skotnicka, A.; Szefler, B.; Kabatc-Borcz, J.; Sutkowy, P. Design and Synthesis of New 5-Methylisatin Derivatives as Potential CDK2 Inhibitors. Int. J. Mol. Sci. 2025, 26, 2144. https://doi.org/10.3390/ijms26052144
Czeleń P, Skotnicka A, Szefler B, Kabatc-Borcz J, Sutkowy P. Design and Synthesis of New 5-Methylisatin Derivatives as Potential CDK2 Inhibitors. International Journal of Molecular Sciences. 2025; 26(5):2144. https://doi.org/10.3390/ijms26052144
Chicago/Turabian StyleCzeleń, Przemysław, Agnieszka Skotnicka, Beata Szefler, Janina Kabatc-Borcz, and Paweł Sutkowy. 2025. "Design and Synthesis of New 5-Methylisatin Derivatives as Potential CDK2 Inhibitors" International Journal of Molecular Sciences 26, no. 5: 2144. https://doi.org/10.3390/ijms26052144
APA StyleCzeleń, P., Skotnicka, A., Szefler, B., Kabatc-Borcz, J., & Sutkowy, P. (2025). Design and Synthesis of New 5-Methylisatin Derivatives as Potential CDK2 Inhibitors. International Journal of Molecular Sciences, 26(5), 2144. https://doi.org/10.3390/ijms26052144