
Tear Sampling and Biomarker Discovery: A Robust Workflow for Routine Clinical Applications Using UHPLC-MS/MS and Schirmer Strips

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
2.1. Mass Spectrometry Method Development
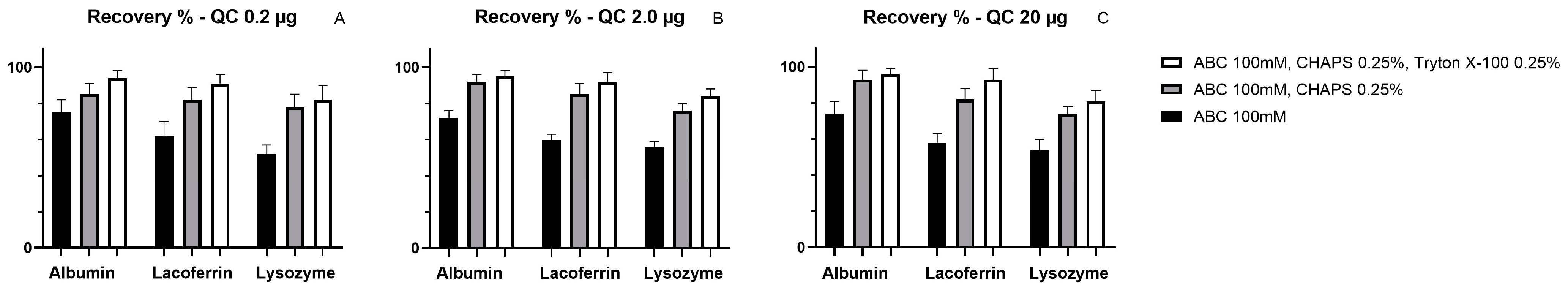
2.2. Extraction Method Development
- Ammonium Bicarbonate (ABC) pH8
- Buffer A: 100 mM ABC pH 8 with 0.25% CHAPS
- Buffer B: 100 mM ABC pH 8 with 0.25% Triton X-100 and 0.25% CHAPS
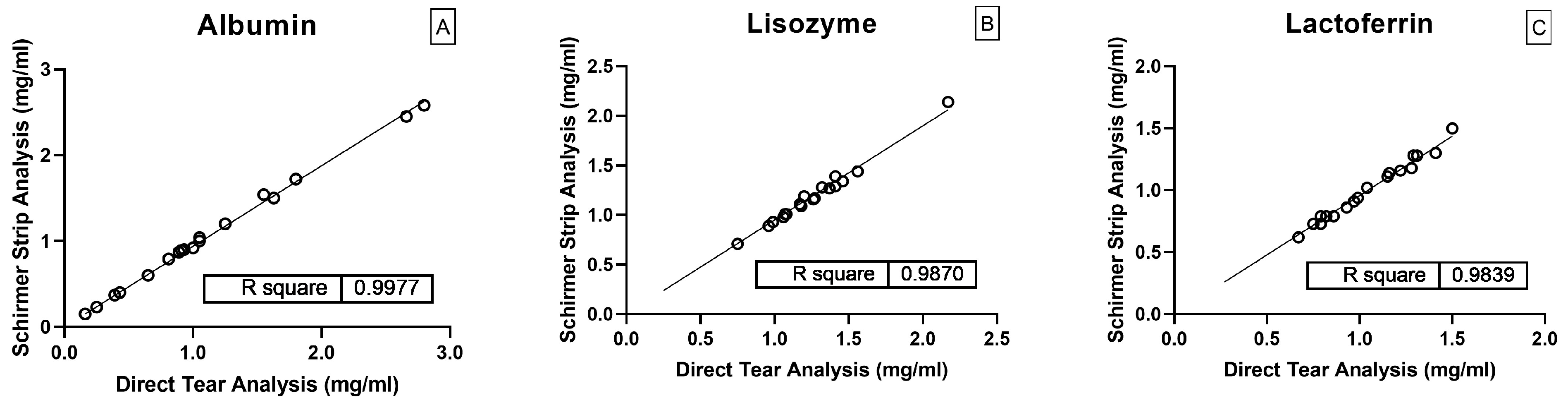
2.3. Method Validation Results
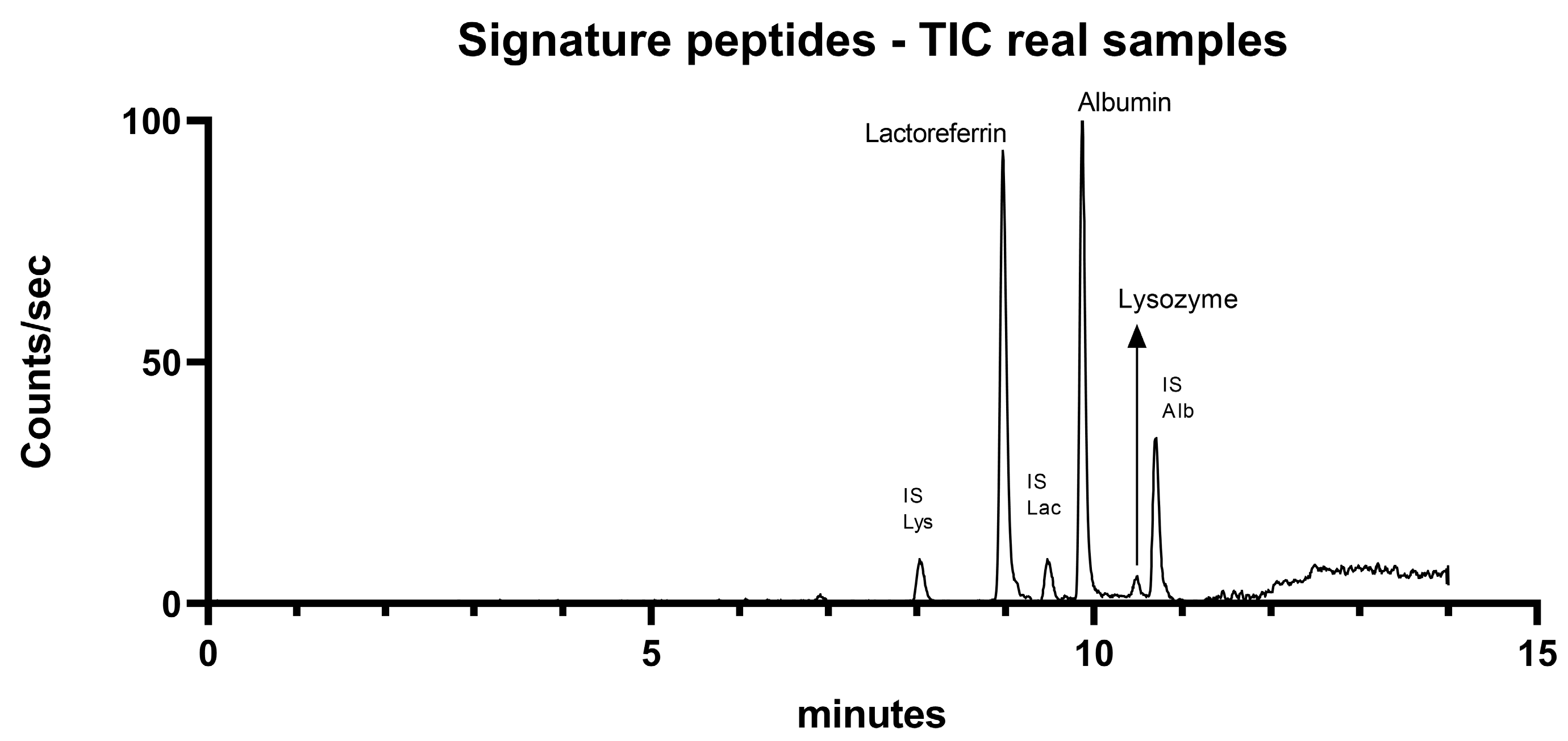
2.4. Application in Real Tear Samples
3. Discussion
4. Materials and Methods
4.1. Reagent and Standard Solution
4.2. Targeted Mass Spectrometry Method for Protein Quantification
4.3. Quality Control Samples and Real Samples
4.4. Extraction Method for Schirmer Strips
4.5. Method Validation
4.6. Electrophoresis for Protein Quantification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2D-SDS PAGE | two-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| LC-MS | liquid chromatography coupled with mass spectrometry |
| ELISA | enzyme-linked immunosorbent assay |
| RPLC | reversed-phase liquid chromatography |
| ESI | electrospray ionization |
| QQQ | triple quadrupole |
| UHPLC | ultra-high-performance liquid chromatography |
| IS | internal standard |
| QCs | quality control |
| MRM | multiple reaction monitoring |
| LOD | limit of detection |
| LOQ | limit of quantification |
| Alb | human albumin |
| Lys | human lysozyme |
| Lact | human lactoferrin |
References
- Dohlman, C.H. The Function of the Corneal Epithelium in Health and Disease. The Jonas S. Friedenwald Memorial Lecture. Investig. Ophthalmol. 1971, 10, 383–407. [Google Scholar]
- Ng, V.; Cho, P.; To, C. Tear Proteins of Normal Young Hong Kong Chinese. Graefes Arch. Clin. Exp. Ophthalmol. 2000, 238, 738–745. [Google Scholar] [CrossRef]
- Posa, A.; Bräuer, L.; Schicht, M.; Garreis, F.; Beileke, S.; Paulsen, F. Schirmer Strip vs. Capillary Tube Method: Non-Invasive Methods of Obtaining Proteins from Tear Fluid. Ann. Anat. Anat. Anz. 2013, 195, 137–142. [Google Scholar] [CrossRef]
- Ohashi, Y.; Dogru, M.; Tsubota, K. Laboratory Findings in Tear Fluid Analysis. Clin. Chim. Acta 2006, 369, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Versura, P.; Bavelloni, A.; Blalock, W.; Fresina, M.; Campos, E.C. A Rapid Standardized Quantitative Microfluidic System Approach for Evaluating Human Tear Proteins. Mol. Vis. 2012, 18, 2526–2537. [Google Scholar] [PubMed]
- Daily, A.; Ravishankar, P.; Harms, S.; Klimberg, V.S. Using Tears as a Non-Invasive Source for Early Detection of Breast Cancer. PLoS ONE 2022, 17, e0267676. [Google Scholar] [CrossRef]
- Kaufmann, Y.; Byrum, S.D.; Acott, A.A.; Siegel, E.R.; Washam, C.L.; Klimberg, V.S.; Mancino, A.T. Proteomic Profiling of Tear Fluid as a Promising Non-Invasive Screening Test for Colon Cancer. Am. J. Surg. 2022, 224, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Nättinen, J.; Aapola, U.; Nukareddy, P.; Uusitalo, H. Clinical Tear Fluid Proteomics—A Novel Tool in Glaucoma Research. Int. J. Mol. Sci. 2022, 23, 8136. [Google Scholar] [CrossRef]
- Chiva, A. Department of Clinical Chemistry, University Emergency Hospital, Bucharest, Romania Tear Biomarkers in Dry Eye Disease. Eur. Ophthalmic Rev. 2019, 13, 21. [Google Scholar] [CrossRef]
- Dor, M.; Eperon, S.; Lalive, P.H.; Guex-Crosier, Y.; Hamedani, M.; Salvisberg, C.; Turck, N. Investigation of the Global Protein Content from Healthy Human Tears. Exp. Eye Res. 2019, 179, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Fullard, R.J.; Snyder, C. Protein Levels in Nonstimulated and Stimulated Tears of Normal Human Subjects. Invest. Ophthalmol. Vis. Sci. 1990, 31, 1119–1126. [Google Scholar] [PubMed]
- Nandi, S.K.; Singh, D.; Upadhay, J.; Gupta, N.; Dhiman, N.; Mittal, S.K.; Mahindroo, N. Identification of Tear-Based Protein and Non-Protein Biomarkers: Its Application in Diagnosis of Human Diseases Using Biosensors. Int. J. Biol. Macromol. 2021, 193, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Koduri, M.A.; Prasad, D.; Pingali, T.; Singh, V.K.; Shanbhag, S.S.; Basu, S.; Singh, V. Optimization and Evaluation of Tear Protein Elution from Schirmer’s Strips in Dry Eye Disease. Indian J. Ophthalmol. 2023, 71, 1413. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, J.; Guo, Y.; Golubnitschaja, O. Mass Spectrometry Analysis of Human Tear Fluid Biomarkers Specific for Ocular and Systemic Diseases in the Context of 3P Medicine. EPMA J. 2021, 12, 449–475. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; D’Alessandro, M.; Di Ioia, M.; Di Ilio, C.; Sacchetta, P.; Del Boccio, P. Unraveling the Molecular Repertoire of Tears as a Source of Biomarkers: Beyond Ocular Diseases. Proteom. Clin. Appl. 2015, 9, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Tham, M.L.; Mahmud, A.; Abdullah, M.; Md Saleh, R.; Mohammad Razali, A.; Cheah, Y.K.; Mohd Taib, N.; Ho, K.L.; Mahmud, M.; Mohd Isa, M. Tear Samples for Protein Extraction: Comparative Analysis of Schirmer’s Test Strip and Microcapillary Tube Methods. Cureus 2023, 15, e50972. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Couchman, L.; Taylor, D.R.; Sandhu, J.K.; Slingsby, O.C.; Ng, L.L.; Moniz, C.F.; Jones, D.J.L.; Maxwell, C.B. Use of Nonhuman Sera as a Highly Cost-Effective Internal Standard for Quantitation of Multiple Human Proteins Using Species-Specific Tryptic Peptides: Applicability in Clinical LC-MS Analyses. J. Proteome Res. 2024, 23, 3052–3063. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting Biomolecular Knowledge Via a Protein Database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.-Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef]
- Lépine, M.; Zambito, O.; Sleno, L. Targeted Workflow Investigating Variations in the Tear Proteome by Liquid Chromatography Tandem Mass Spectrometry. ACS Omega 2023, 8, 31168–31177. [Google Scholar] [CrossRef]
- Ke, X.; Chen, Q.; Pan, X.; Zhang, J.; Mo, W.; Ren, Y. Quantification of Lactoferrin in Breast Milk by Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry with Isotopic Dilution. RSC Adv. 2016, 6, 12280–12285. [Google Scholar] [CrossRef]
- Mörtstedt, H.; Kåredal, M.H.; Jönsson, B.A.G.; Lindh, C.H. Screening Method Using Selected Reaction Monitoring for Targeted Proteomics Studies of Nasal Lavage Fluid. J. Proteome Res. 2013, 12, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lai, S.; Cai, Z.; Chen, Q.; Huang, B.; Ren, Y. Determination of Bovine Lactoferrin in Dairy Products by Ultra-High Performance Liquid Chromatography–Tandem Mass Spectrometry Based on Tryptic Signature Peptides Employing an Isotope-Labeled Winged Peptide as Internal Standard. Anal. Chim. Acta 2014, 829, 33–39. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Signature Peptide | Precursor Ion | Product Ion | Collision Energy | Retention Time (min) |
|---|---|---|---|---|---|
| Albumin | LVNEVTEFAK | 575.3 | 595.3 [y5] *; 694.4 [y6] | 25; 20 | 9.85 |
| IS Albumin | LVNELTEAFAK | 582.3 | 708.4 [y6] *; 837.4 [y7] | 25;20 | 10.28 |
| Lactoferrin | LRPVAAEVYGTER | 487.6 | 462.2 [y4] *; 737.4 [b7] | 20;20 | 8.80 |
| IS Lactoferrin | LRPVAAEIYGTK | 659.3 | 737.4 [b7] *; 850.5 [b8] | 20;20 | 9.52 |
| Lysozyme | STDYGIFQINSR | 700.8 | 764.4 [y6] *; 304.1 [b3] | 25;25 | 10.11 |
| IS Lysozyme | FESNFNTQATNR | 714.8 | 1152.5 [y10] *; 804.3 [y7] | 25;25 | 8.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comito, R.; Ciavarella, C.; Astolfi, G.; Conti, M.; Porru, E.; Violante, F.S.; Versura, P. Tear Sampling and Biomarker Discovery: A Robust Workflow for Routine Clinical Applications Using UHPLC-MS/MS and Schirmer Strips. Int. J. Mol. Sci. 2025, 26, 2041. https://doi.org/10.3390/ijms26052041
Comito R, Ciavarella C, Astolfi G, Conti M, Porru E, Violante FS, Versura P. Tear Sampling and Biomarker Discovery: A Robust Workflow for Routine Clinical Applications Using UHPLC-MS/MS and Schirmer Strips. International Journal of Molecular Sciences. 2025; 26(5):2041. https://doi.org/10.3390/ijms26052041
Chicago/Turabian StyleComito, Rossana, Carmen Ciavarella, Gloria Astolfi, Matteo Conti, Emanuele Porru, Francesco Saverio Violante, and Piera Versura. 2025. "Tear Sampling and Biomarker Discovery: A Robust Workflow for Routine Clinical Applications Using UHPLC-MS/MS and Schirmer Strips" International Journal of Molecular Sciences 26, no. 5: 2041. https://doi.org/10.3390/ijms26052041
APA StyleComito, R., Ciavarella, C., Astolfi, G., Conti, M., Porru, E., Violante, F. S., & Versura, P. (2025). Tear Sampling and Biomarker Discovery: A Robust Workflow for Routine Clinical Applications Using UHPLC-MS/MS and Schirmer Strips. International Journal of Molecular Sciences, 26(5), 2041. https://doi.org/10.3390/ijms26052041